
Total Internal Reflection Fluorescence Microscopy
Basic Microscope Configuration
A wide spectrum of optical configurations was placed under scrutiny during the early stages of instrument development for total internal reflection fluorescence microscopy investigations (TIRFM). From this effort emerged a number of designs that satisfy the requirements for generating a thin evanescent field at the junction between two materials of differing refractive index.
The majority of these designs centered on inverted microscopes, primarily due to the convenience of adding TIRFM optics above, rather than below, the bulky microscope stage. Upright microscope configurations can also be utilized, especially when this represents the only practical choice for the experimental conditions or the investigator's budget. Observations of cell-substrate contacts with cells growing in monolayer culture on the bottom of a plastic Petri dish are a good candidate for TIRFM with an upright microscope, especially when water immersion objectives with a dipping cone are employed. Inverted microscopes are more efficient when examining cells or membrane components attached to the upper portion of a sealed specimen chamber or when the objective is utilized both to illuminate and retrieve secondary fluorescence emission from the specimen.
Regardless of the basic microscope design, a majority of the currently utilized TIRFM configurations rely on an added prism to direct laser illumination toward the interface where total internal reflection occurs, which is in the specimen conjugate plane of the microscope. It is also possible to use the microscope objective to simultaneously direct illumination to the interface and capture secondary fluorescence emission produced by excited fluorophores. This discussion reviews many of the basic microscope configurations for TIRFM, and assumes that the example specimen consists of cells in tissue culture labeled with one or more fluorescent dyes and growing in a monolayer attached to a glass coverslip at the total internal reflection interface.
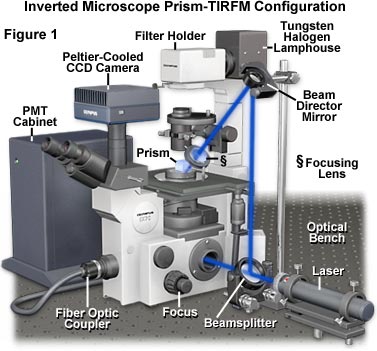
Presented in Figure 1 is a typical TIRFM instrument configuration involving an inverted tissue culture microscope, a trapezoidal prism block, external laser illumination, and a photomultiplier/CCD combination detector system. Blue light emitted from the laser first passes through a beam expander and then into a beamsplitter that divides the light between a microscope entrance port and the beam director mirror. After reflecting from the surface of the director mirror, a portion of the light is focused by the focusing lens onto the trapezoidal prism positioned on the microscope stage just above the specimen chamber and objective. The prism directs illumination to the TIRF interface at an angle that is slightly larger than the critical angle for total internal reflection, creating an evanescent field that excites fluorophores in the specimen. Another portion of the light passed by the beamsplitter enters the microscope port, where it can be directed through the optical train to the specimen for widefield epi-fluorescence excitation. Secondary fluorescence emitted by the specimen is collected by the objective and passed either to the eyepieces, the CCD camera system, or the photomultiplier (PMT). Because the top of the trapezoidal prism is flat, illumination from the tungsten halogen lamphouse can be utilized to observe the specimen in transmitted mode utilizing a variety of contrast enhancing techniques, including differential interference contrast, phase contrast, darkfield, and Hoffman modulation contrast.
When designing TIRFM experiments, the most important factor to consider in the configurational motif is the critical angle of illumination. Ordinary glass coverslips, upon which tissue culture cells are commonly grown, have a refractive index of about 1.52 while the medium surrounding the cells and the interior cellular components have refractive indices ranging from 1.33 to 1.38. Therefore, to obtain total internal refection at the interface between a cell membrane and the coverslip (n(2)/n(1) = 1.52/1.38), the angle of incidence must be larger than the critical angle of 65 degrees. If the cells are not intact and have been permeabilized, hemolyzed, sonicated, or fixed, then the lower refractive index is that of aqueous buffer (n(1) = 1.33) instead of cytoplasm, and the critical incidence angle is reduced to 61 degrees.
In principle, it is possible to employ conventional tungsten halogen or mercury/xenon arc lamps to conduct TIRFM experiments, but a majority of the investigations reported in the literature are performed under laser illumination. The primary reason that lasers are so widely preferred is that laser light is coherent, polarized, and well collimated, so that it can be easily directed into the objective or prism using standard beam expanders, mirrors, and focusing lenses. When the laser source is properly aligned, the total internal reflection interface is illuminated with an area of defined geometry that can be easily adjusted to accommodate experimental variations. High intensity laser sources are also important for experiments that require a significant amount of illumination, such as fluorescence recovery after photobleaching (FRAP) investigations and fluorescence ratio determinations.
The most popular laser sources for TIRMF experiments are the 1 to 3 watt air and water cooled continuous wave argon-ion systems that have strong emission lines at 488 and 514 nanometers, providing the ability to excite a wide spectrum of common fluorophores. Lasers having lines at longer wavelengths, such as helium-neon and krypton-ion gas lasers, can be employed to excite fluorophores that absorb in the 540 nanometer and higher regions. However, virtually any laser with a total visible output in the 0.5 watt or greater range should be adequate. A laser source is generally preferable to an arc lamp for TIRFM, because collimation of the light emitted by the arc lamp results in severe reduction of intensity. However, laser illumination suffers from unavoidable interference fringing on the specimen, which can be somewhat reduced by meticulously cleaning the optical surfaces. For critical experiments, investigators should explore rapid jiggling of the laser beam or at least compute a normalization of sample digital images using a control digital image consisting of a uniform concentration of fluorophores.
Inverted Microscope Configurations
The simplest approach to achieve total internal reflection from a culture chamber on an inverted microscope is direct laser illumination through a glass cube positioned on top of the chamber, as illustrated in Figure 2. In general, a fixed-stage microscope (in which the nosepiece/objective combination is translated during focusing) is more convenient than a movable stage microscope. This configuration ensures that the specimen will remain stationary during focusing, requiring only an initial alignment of the laser beam. However, moving-stage microscopes are more common and either type can be employed for these experiments.
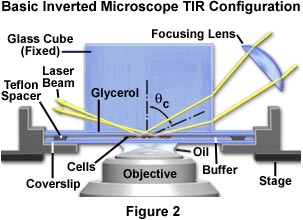
In all inverted microscope configurations considered here, the buffer-filled sample chamber consists of a lower bare glass coverslip, a 60-micron thick Teflon spacer ring, and the cell adhesion coverslip inverted so the cells face downward towards the microscope objective. An advantage of this design is that cells not bound to the substrate, loose cellular debris, and contaminating protein tend to sediment as an aggregate to the bottom of the chamber without obscuring or adding extraneous fluorescence to the total internal reflection interface.
The upper surface of the cell monolayer-coverslip combination is placed in optical contact with the glass block (or prism) by means of a thin layer of immersion oil or glycerol. The lateral faces of the glass block should be fixed, but the sample must be able to undergo translation in the x and y directions while still maintaining optical contact. The lower coverslip can be oversized and the Teflon spacer is often made with gaps so that the buffer solution bathing the cells can be changed by capillary action with entrance and exit ports (Figures 2 and 3). Several manufacturers offer tissue culture chambers that are designed specifically for this purpose.
As demonstrated in Figure 2, the laser beam first enters a focusing lens positioned obliquely above the microscope stage. The purpose of the lens is to concentrate laser illumination in much the same manner as an objective during epi-illumination, but the lens also serves to narrow the beam width for easier alignment. The focal length of the lens is not critical and can range between 50 and 100 millimeters, but should allow the investigator to easily change the size of the illuminated area over a narrow range. As with other components in the illumination system, the focusing lens should be mounted on an x-y-z translator having one of the axes aligned with the incoming laser beam direction. The translator can be mounted either on the laboratory bench or the microscope stage, but will be easier to align when it is mounted on the stage.
Three primary optical elements control and direct the laser beam size and pathway prior to its entering the focusing lens (see Figure 1). These consist of a beam expander to widen the laser light beam as it exits the cavity, a beamsplitter (optional) or mirror, and a beam director to divert the beam into the focusing lens.
Laser beam expanders are widely available from a number of distributors, and are employed to control the width of the beam at the focusing lens. As a consequence, the angle of convergence onto the specimen and the width of the focused spot is then determined by the focusing lens. Laser light is refracted at a series of angles across the beam profile when it encounters the vertical face of the cube. The resulting geometric aberration produces an elliptically illuminated area at the interface, which is elongated when compared to the expected pattern for a parallel light beam with a Gaussian profile. Beams that are wider when they enter the focusing lens produce a reflection spot having thinner dimensions. Under optimum conditions, a beam half-width approaching a lower limit of 2.5 microns can be attained.
The beamsplitter is a mirror that diverts a horizontal laser beam into a vertical direction (at a 90-degree angle) to elevate it past the height of the microscope stage and glass prism or block. An adjustable optical mount should be utilized to house the beamsplitter so that it can be removed to allow direct entry of laser light into a microscope port for quick and reversible conversion to auxiliary epi-illumination. By using a partially silvered mirror or even a plain glass slide as a beamsplitter, both epi-illumination and total internal reflection can be viewed simultaneously.
| Interactive Tutorial | |||||||||||
|
|||||||||||
A beam director mirror is employed to angle the laser beam from the vertical direction obliquely down toward the focusing lens (see Figure 1). The mirror height determines the incidence angle at the total internal reflection surface. In addition to the linear height adjustment, the director mirror should be mounted on a biaxial rotational mount to allow direction of the unfocused laser beam into the prism. If the mirror mount is attached to the stage of a moving-stage microscope (rather than the laboratory bench), the position of the reflection region can be rendered insensitive to changes when focusing the specimen.
When properly aligned, the collimated and focused laser beam enters the glass cube, and is directed by refraction to strike the total internal reflection interface (beneath the cube's lower surface) with an incident angle exceeding the critical minimum. Neither the size nor shape of the cube is vital, and a prism or rectangular glass block can easily be substituted. Equilateral and 45-45-90-degree prisms are standard commercial items and can be purchased from a variety of sources. A prism with a flat top (Figures 2 and 4), such as the cube described above or a truncated triangle, allows placement of a tungsten halogen lamp and condenser system above the prism. In this configuration, conventional illumination techniques such as brightfield, darkfield, phase contrast, and differential interference contrast can be coupled to TIRFM investigations to assist in determining the spatial location of fluorescence originating at the interface.
To install the prism or glass block, apply a droplet of pure glycerol or immersion oil to the upper surface of the culture chamber and slowly lower the prism onto the oiled surface, thereby spreading the droplet into a thin layer. The contact media (glycerol or oil) serves two purposes: to ensure optical contact between the prism and the specimen chamber, and to mechanically lubricate the region between the two glass surfaces. As the specimen chamber is laterally translated during scanning, the prism or glass block remains fixed. The incident laser beam should propagate obliquely through the lower portion of the prism, then through the glycerol or oil layer, and finally through the specimen, totally reflecting at the bottom surface of the substrate directly over the optical axis of the microscope. Substrate thickness is not critical, except that thick substrates (those approaching a millimeter or even more) may constrain the laser beam from meeting the bottom surface directly over the microscope's optical axis.
The immersion contact media (oil or glycerol) should be used sparingly, because any excess may accumulate at the lower edges of the prism and interfere with the incident laser illumination when it first enters the glass. Prism size is not critical, but larger prisms may inhibit lateral motion of the specimen, although they will increase the clearance between the immersion media bead and the incident light beam. In general, a prism or glass block with a face area of about a square centimeter is ideal.
The prism employed to couple exciting laser light into the system does not require an exact match in refractive index to the slide or coverslip in which total internal reflection takes place. They may be optically coupled with glycerol, cyclohexanol, or microscope immersion oil, among other optically distinct liquids. Immersion oil has a higher refractive index, which helps to avoid possible total internal reflection at the prism/coupling media interface at low incident angles. However, many brands of immersion oil demonstrate a significant amount of autofluorescence. Another important point is that a truncated prism surface does not require special polishing treatment, but will perform adequately with the smoothness of a standard commercial microscope slide.
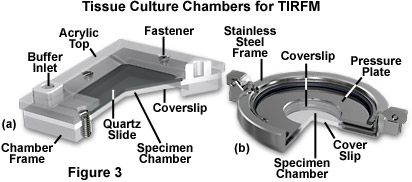
Several specimen chamber designs that utilize windows composed of glass slides and/or coverslips are presented in Figure 3. On the left (Figure 3(a)) is a Tamm chamber devised to provide a thin profile that allows the tissue culture cells to be observed through a thin layer of buffer solution, which permits the use of high numerical aperture objectives having very short focal lengths. A monolayer of cells, grown on the upper glass coverslip (or microscope slide), are separated from the lower coverslip with a thin spacer having a thickness less than 100 micrometers. Access holes enable the cells to be perfused with a variety of solutions to conduct titrations, equilibrium binding experiments, or kinetic analysis of interactions at the cell surfaces. A similar device, the Dvorak-Stotler controlled-environment culture chamber, is illustrated in Figure 4(b). This chamber also utilizes coverslips for both the top and bottom windows, but does not have access holes or any mechanism to allow addition of chemicals or changes to the buffer solution.
As depicted in Figure 3, the specimen chambers contain the aqueous buffer solution necessary to maintain the cultured cells. This solution is sandwiched between the substrate surface and a glass coverslip, which are separated by a Neoprene or Teflon spacer. Teflon rings are commercially available, having thicknesses ranging upwards from 50 micrometers. The buffer solution depth must be quite thin if high-magnification objectives having large numerical apertures and short working distances are to be employed. In many cases, the downward pressure of the glass prism upon the substrate plate may be adequate to seal the specimen chamber without additional clamping, but many chamber designs (Figure 3) include mechanisms to secure the substrate and glass coverslip.
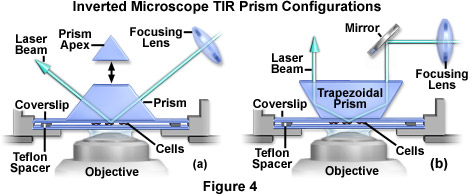
In cases where a prism is employed (as opposed to a glass block) to achieve total internal reflection (see Figure 4(a)), the maximum incidence angle is obtained by introducing the laser beam from the horizontal direction. For standard glass (having a refractive index of 1.52), the maximum incidence angle is 73 degrees for a right angle prism and 79 degrees for an equilateral prism. Phase contrast and other transmitted light techniques are not compatible with this configuration because the upper surface of the triangular prism is not flat. However, custom truncation and polishing of the prism top (presented as an option in Figure 4(a)) produces a surface that can easily pass incident light from above by an inverted microscope condenser system.
| Interactive Tutorial | |||||||||||
|
|||||||||||
When mounted on the microscope condenser unit, a 60-degree trapezoidal prism (Figure 4(b)) is the most convenient and reproducible configuration yet developed for TIRFM above the stage on an inverted instrument. The incoming laser beam is vertical, so the total internal reflection area shifts laterally to a very small degree when the prism is raised and relowered during specimen changes. In addition, conventional transmitted light techniques (phase contrast, brightfield, etc.) are compatible with this experimental design. Because the incident angle is fixed at 60 degrees, ordinary optical glass (refractive index of 1.52) is not able to support total internal reflection, and a prism having a high refractive index is required. Prisms fabricated with flint glass (refractive index of 1.64) will meet these specifications, and are commercially available. The beam will then refract away from the normal at an angle of 69 degrees in passing from the prism into the coverslip, thereby exceeding the critical angle at the coverslip/buffer or coverslip/cell interface. A trapezoid with walls ranging between 45 and 60 degrees is ideal, but these units are not readily available and must be manufactured to custom specifications. Unfortunately, 45 or 60-degree trapezoids are also not commercially available, but they can be cheaply produced by truncating and polishing the apex of a commercially available triangular prism.
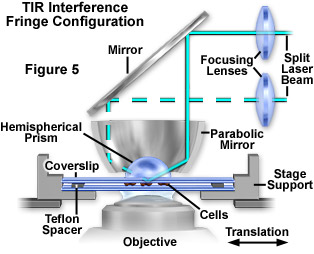
Presented in Figure 5 is a fourth configuration utilizing a parabolic mirror and hemispherical prism to conduct TIRFM experiments with an inverted microscope. In this design, the mirror and prism are positioned so that the laser beam traverses a radius of the prism toward a total internal reflection target area at the focus of the parabolic mirror. Because the light rays enter the prism almost normal to the face at all incidence angles, aberration of the beam profile is reduced compared to that observed with a cube or rectangular prism block. A lateral shift of the vertical incoming laser beam will always focus at the same spot, enabling substantial and convenient changes to the incident light angle. In addition, interference fringes in the evanescent field can be created by splitting the incoming beam into two components, each reflecting at a separate azimuthal position in the parabola, but recombining at the parabolic focal point. The fringe spacing can be varied by adjusting the relative azimuthal positions of the two incident beams, and very high spatial frequencies can be attained. Interference fringes can be employed as an aid to focusing on the interface by creating a parallel line interference fringe pattern. This pattern can be observed in fluorescent regions of cell/substrate contact, which confirms the excitation source as the evanescent field (rather than light randomly scattered from the field by cells). Interference patterns are also useful in studies of surface diffusion rates to measure the lateral mobility of membrane components in contact regions. Despite the versatility of this design (Figure 5), alignment is very difficult and the system is more sensitive to vibrations.
The inverted microscope TIRFM configurations reviewed here all share some advantages and disadvantages. Among the major advantages are the ability to utilize rather inexpensive optics with ample room on the microscope stage to position the specimen and glass block/prism and supporting optical components. In addition, the prism may be mounted on the condenser holder (housed above the stage on modern inverted microscopes) for ease in raising and lowering, and for auxiliary observation with conventional optical techniques. Finally, these optical arrangements enjoy simplicity in design and are the easiest systems to align for total internal reflection.
Among the chief disadvantages of inverted microscope configurations is the fact that the specimen is not easily accessible from above and conventional illumination is hampered by the prism apex. Also, the shortest working distance objectives may not reach focus across the buffer layer, and image quality with high numerical aperture objectives is reduced due to spherical aberration when viewing specimens through the relatively thick buffer layer.
Alignment of inverted microscope systems with a prism or glass block involves centering the total internal reflection region directly over the objective while the specimen is in sharp focus. The first step is to assemble the specimen chamber and prism combination, then bring the specimen into focus using brightfield illumination (even in cases where the prism apex is present). Next, with the laser-focusing lens removed, direct the beam so that it undergoes total internal reflection over the center of the objective front lens. This can be observed by looking directly (not through the microscope eyepieces) for scattered incident light in the prism, immersion layer, and substrate plate. After re-inserting the focusing lens, adjust its position so that the focused laser beam enters the prism side surface approximately one millimeter above the bottom edge. Directly viewing the beam should reveal three co-linear spots of scattered light at the bottom of the prism: two on the outside where the beam traverses the immersion layer, and a central spot where the beam undergoes total internal reflection on the specimen. The focusing lens should be adjusted so the middle spot is visible as an elongated cylindrical area. The position and size of the total internal reflection region can be adjusted with the focusing lens translators and by moving the lens longitudinally to optimize the illumination region dimensions.
The shape of the total internal reflection area is dependent upon the orientation of the prism or glass block side surface, where the laser beam enters. If the side is vertical (as in a block or rectangle), the area will be elongated to a degree that is a function of beam convergence. As previously discussed, elongation is due to aberration introduced by the large refraction angle at the first surface of the prism or glass block. On the other hand, if the side surface is angled so the collimated laser beam enters at an angle close to the perpendicular, then the reflection area will resemble a less-elongated conical section, which is expected from slicing a cylindrical beam at an angle.
In all of the optical configurations described above, the illuminated region will remain stationary in the center of the viewfield as the specimen is scanned in the lateral dimensions. However, when the microscope stage is moved during focus, the illuminated region may move laterally as the specimen is focused, depending upon whether the focusing lens and beam director are mounted on the laboratory bench or the microscope stage.
The total internal reflection spot should be focused to a width no larger than the field of view. If the spot is too large, spurious scattering and out-of-focus fluorescence from the immersion oil layer between the prism and coverslip will increase the otherwise low fluorescence background that is possible with TIRFM. Another artifact occurs at incident angles very near the critical minimum, which is manifested by a noticeable shadow cast by the cells along the surface of the interface. This can be avoided by making certain the incidence angle exceeds the critical angle by several degrees.
When the incident angle is near the critical value, it is sometimes difficult to ascertain whether total internal reflection has been achieved. In this case, viewing the resulting fluorescence intensity through the microscope eyepieces can often help the investigator determine if the instrument is properly configured. Fluorescence emission from a TIRF interface is quite distinct from that excited by an incident beam propagating at a subcritical "skimming" angle through the aqueous buffer. Because the evanescent wave (unlike the propagating beam) is far shallower than the objective depth of focus, fluorescence appears to originate from a single plane when excited by total internal reflection. In contrast, epi-illumination (from a subcritical beam) excites fluorophores throughout the bulk of the specimen chamber and secondary fluorescence is observed over a wide range of focal planes.
| Interactive Tutorial | |||||||||||
|
|||||||||||
For local qualitative intensity measurements in total internal reflection experiments, it may be desirable to limit the area from which emission light is gathered. Often the illuminated area is not likely to be very small, but a defined surface area can be obtained with an adjustable diaphragm strategically placed at an image plane in the emission light pathway.
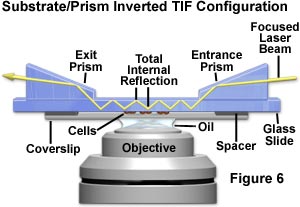
Several additional TIRFM configurations involving inverted microscopes have been employed by investigators. The system illustrated in Figure 6 presents an alternative system that fixes the prisms with respect to the specimen instead of the laser beam. Illuminating laser light enters from the right through an entrance prism, and is refracted into the substrate where it propagates toward the microscope's optical axis via multiple internal reflections. Along the way, the multiply reflected beam encounters the specimen (tissue culture cells adhering to the glass coverslip) and illuminates chromophores in the region of the glass/buffer interface. After passing through the specimen and substrate, the reflected beam passes into the exit prism, which directs the light away from the experiment. In this configuration, the illuminated reflection area will move when the specimen is translated.
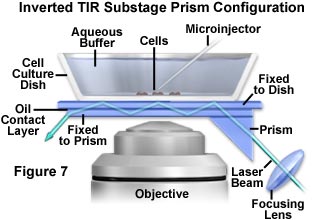
A more useful configuration that is compatible with simultaneous microinjection or patch clamp experiments is presented in Figure 7. In order to provide easy and continuous access from above to a tissue culture bathed in buffer, the prism is deployed below stage level to contact the substrate glass. This configuration, which cannot be employed for all inverted microscopes, suffers from tight geometry because the objective also resides in the immediate area. Like the system illustrated in Figure 6, the substage prism initiates multiple total internal reflection in the coverslip, which transfers excitation light as a waveguide from far off-axis to the center of the viewfield.
In the simplest form, a small triangular prism (commercially available) is placed, via immersion oil or glycerol, in optical contact with the bottom of the coverslip containing the cells and buffer solution. The specimen can be translated while the prism remains laterally fixed, although a smear of oil may be left on the lower side of the coverslip that can destroy the first internal reflection (and the experiment). A feasible alternative is to employ an additional intervening coverslip fixed to the prism with optically transparent glue. The sliding motion occurs between the intervening coverslip and the cell coverslip, which are in optical contact and lubricated by a thin layer of immersion oil or glycerol. A major disadvantage of this configuration is that oil or glycerol immersion objectives cannot be used because the immersion medium could destroy the internal reflections prior to formation of the illumination spot. However, air (dry) or water immersion objectives work very well under these circumstances.
Upright Microscope Configuration
A trapezoidal prism, similar to the one illustrated in Figure 4(b), can be mounted on the condenser support frame in an upright microscope to observe phenomena at the total internal reflection interface of specimens positioned on the stage (Figure 8). The expanded laser beam is introduced into the microscope base using the same port intended for the transmitted light illuminator (which should be removed). A long focal length (approximately 250 millimeters) converging lens placed near the entrance port and mounted on an x-y translator is utilized to focus the incoming laser beam and allow fine-tuning of the lateral position. Collimated laser illumination can then take advantage of the microscope's built-in optical train to direct the beam through the base and vertically upward into the prism. An extra lens positioned just above the microscope base is often useful to translate and focus the total internal reflection spot. This configuration does not offer a great deal of flexibility in adjusting the incidence angle, but a limited range of values can be achieved by employing several trapezoids having differing angular geometries and refractive indices. Images are collected through the cooled scientific CCD digital camera illustrated in Figure 5, but a photomultiplier or avalanche photodiode could easily be substituted.

Choice of prism glass is limited by refractive index requirements, with flint glass (refractive index of 1.62) being the preferred formulation. With a 60-degree incidence angle, the refractive index of the prism must be at least 1.53 in order for total internal reflection to occur at the glass/water interface (regardless of the substrate material). This is why flint glass, rather than ordinary optical (crown) glass or fused silica, is the ideal prism material for these experiments.
The illuminating laser light is reflected into the prism by the substage microscope mirror, and refracted through the glass by the angular sides of the prism. Because trapezoidal prisms cut in custom dimensions are not commercially available, the rhombic cross-section is manufactured by truncating and polishing the top of an easily obtainable equilateral triangle prism. The focusing lens is removed for positioning of the laser trajectory, which is adjusted to enter the bottom of the prism off-center where it totally internally reflects at the sloping side and proceeds toward the upper surface at an incidence angle of 60 degrees. When the focusing lens is inserted into the light path, the beam follows the same course but is much thinner and more intense.
The specimen chamber, which may be a plastic culture vessel or Petri dish, is placed on the microscope stage for observation. A small drop of immersion medium (glycerol or oil) is placed on the prism top, and it is brought into optical contact with the lower surface of the culture chamber by raising the substage condenser rack mechanism. The laser beam position is then adjusted by the focusing lens translator to totally internally reflect at the upper surface of the chamber (or substrate) and follow a symmetrical course back down toward the microscope base on the opposite side of the prism. This setup is suitable for many types of substrate, but is particularly well-adapted for viewing cells growing on the bottom of plastic culture dishes. The cells may then be viewed in the same chamber in which they were originally seeded and incubated without any remounting step. Observing the specimen in a upright microscope enables the investigator to simultaneously probe the cells with micropipettes, patch clamps, and injection syringes, provided the objective has a sufficient working distance.
Secondary fluorescence emission may be collected by a dry, oil, or water immersion objective (after decanting most of the buffer) through a coverslip that is suspended above the cell monolayer surface by a spacer. Alternatively, a water immersion objective with a dipping cone can be placed directly into the buffer solution bathing the cells. The upright microscope configuration is very convenient, because specimens may be interchanged easily as they are with standard illumination systems. The position of the TIRF illumination area is also stationary and unaffected by focusing, even on a moving-stage microscope, which accounts for a vast majority of modern upright microscope designs. The chief disadvantage is that the incident illumination angle is not adjustable without switching to another prism with a different slope angle.
An upright system provides high quality images when water immersion objectives are submerged to image cells directly through the buffer solution in an uncovered Petri dish or culture chamber, but the configuration does present several drawbacks. Many of the polymer formulations used in culture vessel construction have some degree of autofluorescence, which may reduce contrast in weakly fluorescent specimens. This effect can be compounded if the immersion-coupling medium is not carefully chosen. Several commercial brands of immersion oil also display autofluorescence at varying levels, and investigators are cautioned to examine both immersion oils and plastic culture vessels for potential fluorescence artifacts before they are used in TIRFM experiments. Different brands of tissue culture plastic have significant variations in the amount of autofluorescence displayed upon excitation.
The prism, slide, chamber windows, and coverslips can be made with ordinary flint optical glass for most applications, unless shorter penetration depths arising from higher refractive indices are desired. Optical glass does not transmit light below about 310 nanometers and also has a dim autoluminescence with a long decay time, which can be a problem in some photobleaching experiments. The autoluminescence of high quality fused silica (quartz) is much lower. More exotic high refractive index materials, such as sapphire, titanium dioxide, and strontium titanate, do not suffer from high levels of autofluorescence and yield evanescent field penetration depths more than an order of magnitude lower than the illumination wavelength.
Detection of Fluorescence
Unlike confocal and multiphoton microscopy, many TIRFM images can be recorded on film in a manner similar to the classical forms of microscopy that rely heavily on photomicrography (brightfield, darkfield, phase contrast, differential interference contrast, etc.). In some cases, especially when only a few fluorophores are present in the specimen, it is advisable to use more sensitive imaging devices, such as a intensity-enhanced video camera, cooled scientific CCD, avalanche photodiode, or a photomultiplier. Time-lapse cinemicrography is also possible in TIRFM experiments, and the sequence of image frames can be captured with any of the devices mentioned above, the choice of which will depend upon the intensity of secondary fluorescence and the exposure times necessary to acquire images. Richly fluorescent specimens can be recorded on film (singly or in a sequence of frames), but image acquisition with a video or CCD camera is far more convenient with the current hardware readily available to investigators.
A significant amount of versatility is afforded when images are acquired with a photomultiplier tube or avalanche photodiode. Photomultipliers are often employed for spatially unresolved but fast detection and quantitation of fluorescence intensity. Variable apertures and/or image plane diaphragms (discussed above) enable the investigator to selectively capture only portions of the fully available field. This is useful when long sampling intervals at low-light levels are necessary, or when only part of the viewfield is of interest. The photomultiplier spectral characteristics should be considered when choosing a device for image collection, and these should coincide with the emission properties of the fluorophores to be examined.
Conclusions
In general, total internal reflection is more difficult to implement on a microscope that has a moveable stage because focus adjustments require realignment of the laser beam into the focal plane and optical axis of the microscope. When choosing a microscope for TIRFM investigations, pay attention to the mechanical stability and construction motif of the instrument. Inverted or upright microscopes with a heavy frame on an isolation table will enable the investigator to establish a relatively vibration-free environment for the fragile evanescent field. Maintaining a focused laser illumination source within a range of a few microns over long periods is difficult (at best), but can be accomplished when the microscope, laser source, and supporting optical components are secured in proper mounts and firmly fastened to the bench or microscope stage.
A wide range of microscope objective designs are compatible with TIRFM experiments. The only absolute requirement is that the objective have a long enough working distance to observe the interface across a thin gap of buffer solution defined by the spacer thickness. Teflon and Neoprene spacers can be made thinner than any standard objective working distance, while still being much larger than the penetration depth of the evanescent field. This reduces the likelihood of being unable to focus on the interface, but does not compensate for potential aberrations resulting from refractive index variations in the specimen/buffer region. Correction of spherical aberration in most high-magnification objectives is critically dependent upon the specimen being immediately on the far side of a coverslip rather than across an additional thin water layer. The result can be an image lacking in contrast and producing a hazy image even when sharply focused. No general rule for objective specifications can be provided, but water immersion objectives (either dipping or those requiring a coverslip) are probably the best (and most expensive) choice. On the other hand, TIRFM experiments have been successfully conducted with objectives of almost every commercially available numerical aperture and magnification.
During many investigations, it is desirable to switch rapidly between illumination of the interface by total internal reflection and deeper penetration into the bulk of the specimen by classical epi-fluorescence illumination. As an example, a transient process may involve simultaneous, but somewhat different, changes in both the membrane and cytoplasm, both of which must be recorded. Specially designed optoelectronic systems that take advantage of acousto-optic modulators have been described by several investigators. These configurations are computer-controlled and usually contain several modulators that act in synchrony to oscillate illumination between TIRF and epi-fluorescent widefield, while simultaneously collecting data with a photomultiplier or fast-response photodiode device.
The glass surface supporting cell adhesion is often coated with specific substrates for TIRFM investigations. Chemical derivatives of the silicon dioxide surface can yield unique physical and chemical absorption properties that are useful in modulating the formation of model membranes and similar structures. In addition, the covalent attachment of certain specific chemicals is particularly useful in cell biology and biophysics. Cell adhesion can be enhanced by attachment of poly-L-lysine to the glass surface, and hydrophobic surfaces can be created with long-chain hydrocarbons for lipid monolayer absorption. Other useful molecules that can be used to treat the surface for selective specificity include antibodies, antigens, planar phospholipids, and lectins.
Thin layers of aluminum (in the region of 20 nanometers), which can quench fluorescence, can also be applied to the glass surface by standard vacuum evaporation. The deposition thickness can be precisely controlled by completely evaporating a pre-measured constant amount of aluminum. After deposition, the upper surface of the aluminum film spontaneously oxides in the atmosphere to produce a thin layer of aluminum oxide. The oxide-coated film has chemical properties similar to silicon dioxide and can be derivatized by organosilanes in a manner similar to that of glass. The ability of a metal film to almost completely quench fluorescence of those fluorophores within about 10 nanometers allows suppression of the signal from labeled proteins that are nonspecifically absorbed to the substrate. This occurs while permitting fluorescence from the slightly more distant labeled protein adsorbed to an adherent membrane.
Unlike confocal microscopy, which produces optical sections by exclusion of out-of-focus emitted light with a set of image plane pinhole diaphragms, TIRFM is limited to the area within a few hundred nanometers of the glass/buffer interface where total internal reflection is occurring. Confocal microscopy has a clear advantage in versatility because optical sectioning works at any plane of the specimen and is not restricted to an interface between dissimilar refractive indices. However, TIRFM does have several advantages including a much thinner optical section (around 100 nanometer versus 600 nanometers for confocal), selective illumination of the immediate area adjacent to the interface, and the ability to (relatively inexpensively) adapt existing microscopes to take advantage of the technique.
Contributing Authors
Daniel Axelrod - Department of Biophysics, 930 North University Ave., University of Michigan, Ann Arbor, Michigan 48109.
Michael W. Davidson - National High Magnetic Field Laboratory, 1800 East Paul Dirac Dr., The Florida State University, Tallahassee, Florida, 32310.
BACK TO TIR FLUORESCENCE MICROSCOPY