
Multiphoton Fluorescence Microscopy
Introduction
Multiphoton fluorescence microscopy is a powerful research tool that combines the advanced optical techniques of laser scanning microscopy with long wavelength multiphoton fluorescence excitation to capture high-resolution, three-dimensional images of specimens tagged with highly specific fluorophores.

The methodology is particularly useful to cell biologists who endeavor to study dynamic processes in living cells and tissues without inflicting significant, and often lethal, damage to the specimen. Although classical widefield fluorescence microscopy can often provide submicron resolution of biochemical events in living systems, the technique is limited in sensitivity and spatial resolution by background noise caused by secondary fluorescence throughout areas situated above and below the focal plane.
Excitation in multiphoton microscopy occurs only at the focal point of a diffraction-limited microscope, providing the ability to optically section thick biological specimens in order to obtain three-dimensional resolution. Individual optical sections are acquired by raster scanning the specimen in the x-y plane, and a full three-dimensional image is composed by serially scanning the specimen at sequential z positions. Because the position of the focal point can be accurately determined and controlled, multiphoton fluorescence is useful for probing selected regions beneath the specimen surface. The highly localized excitation energy serves to minimize photobleaching of fluorophores attached to the specimen and reduces photodamage, which increases cell viability and the subsequent duration of experiments that investigate the properties of living cells. In addition, the application of near-infrared excitation wavelengths permit deeper penetration into biological materials and reduce the high degree of light scattering that is observed at shorter wavelengths. These advantages enable researchers to conduct experiments on thick living tissue samples, such as brain slices and developing embryos that would be difficult, if not impossible, to image with other microscopy techniques.
Illustrated in Figure 1 is a typical configuration used in multiphoton fluorescence microscopy experiments. The microscope is an inverted-style instrument designed to observe living cells in tissue culture and thicker biological specimens bathed in saline solution. For routine non-fluorescence observation, a diascopic lamphouse is positioned above the stage to enable visualization of specimens through conventional techniques such as brightfield, differential interference contrast, phase contrast, and Hoffman modulation contrast. Although not useful in multiphoton applications, a 35-millimeter camera body is attached to the port at the front base of the microscope depicted in Figure 1 to capture images taken with conventional illumination. To the right of the microscopy body is a Ti(titanium):Sapphire mode-locked pulsed laser system that is one of the preferred sources for multiphoton excitation due to the high peak intensity but low average power. Control of the laser is accomplished through electronic units situated on top of the laser cabinet, which is joined to a microscope port through either fiber coupling with an optical waveguide or direct coupling with strategically placed relay mirrors. A filtered photomultiplier detection system is attached to another port at the microscope base, as is an x-y raster scanning unit that can rapidly deflect the focused laser beam across the objective field. Digital images collected by the microscope are processed and analyzed by an accompanying computer workstation that can assemble three-dimensional reconstructions from optical sections.
Traditional widefield fluorescence microscopy is plagued by secondary fluorescence occurring away from the focal region, which contributes to flair and a high background noise signal, often obscuring important specimen details. Confocal microscopy circumvents this problem, to a large degree, by rejecting out-of-focus background fluorescence through the use of pinhole apertures, which produce thin (less than a micron) unblurred optical sections from deep within thick specimens. The introduction of multiphoton fluorescence microscopy provides a new alternative to confocal microscopy through selective excitation coupled to a broader range of detection choices. Unlike conventional confocal microscopes, the microscope presented in Figure 1 does not require a pinhole near the detector to attain three-dimensional discrimination, dramatically increasing the efficiency of emitted fluorescence signals. In the past, the high cost and complexity of pulsed laser systems required for multiphoton excitation have limited use of the technique, but recently-introduced turnkey lasers and commercial multiphoton systems have made multiphoton fluorescence microscopy the method of choice for many investigations.
Two-Photon and Three-Photon Excitation
The basic principles of multiphoton excitation were first described by Maria G�ppert-Mayer while conducting her doctoral dissertation research over 70 years ago, but the hypothesis could not be confirmed until the invention of pulsed ruby lasers, about 30 years later. At high photon densities, two photons can be simultaneously absorbed (mediated by a virtual state) by combining their energies to provoke the electronic transition of a fluorophore to the excited state. Because the energy of a photon is inversely proportional to its wavelength, the two photons should have wavelengths about twice that required for single-photon excitation. As an example, two photons having a wavelength of 640 nanometers (red light) can combine to excite an ultraviolet-absorbing fluorophore in the 320-nanometer region (ultraviolet), which will result in secondary fluorescence emission of longer (blue or green) wavelengths. This unique application means that longer wavelengths, extending into the infrared region, can be conveniently utilized to excite chromophores in a single quantum event, which subsequently emit secondary radiation at lower wavelengths.
The requirement of two photons for each excitation event necessitates a rate constant that depends upon the square of the excitation intensity. Although the photons do not have to be of identical wavelength to induce multiphoton excitation, most experimental systems are designed with a single laser source, so the two photons are usually members of a defined population having a narrow wavelength distribution. Unlike the case for single-photon absorption, the probability that a given fluorophore will simultaneously absorb two photons is a function of both the spatial and temporal overlap between the incident photons. Calculations based on the assumption that each fluorophore is exposed to the same laser cross section indicate that photons must arrive within 10(-18) seconds (one attosecond) of each other. The time scale of this overlap period is consistent with the lifetime (10(-17) seconds or 0.01 femtosecond) of the intermediate virtual state.
High photon densities are necessary in multiphoton fluorescence to ensure a sufficient level of fluorophore excitation. In fact, photon concentration must be approximately a million times that required for an equivalent number of single-photon absorptions. This is accomplished with high-power mode-locked pulsed lasers, which generate a significant amount of power during pulse peaks, but have an average power that is low enough not to damage the specimen. Brief, but intense, pulses emitted by the laser increase the average two-photon absorption probability for a given fluorophore at a constant average incident laser power level. Minimizing the average excitation power level reduces the amount of single photon absorption, which also occurs in the specimen during excitation. It is the single photon excitation events that lead to a majority of the heating and some of the photodamage that occurs during fluorescence experiments.
Typical pulsed laser configurations employ short duty cycles of around 100 femtoseconds (10 e(-13) seconds) with a repetition rate of 80 to 100 megahertz for multiphoton fluorescence experiments. This regime permits satisfactory image acquisition without subjecting the specimen to an excessive amount of heat and photodamage. The time scale for each pulse, while often referred to as "ultrashort", is still four to five orders of magnitude longer than the reaction time for two-photon absorption. The population of singlet states in chromophores excited by a two-photon pulse is identical to that obtained during conventional widefield or confocal fluorescence microscopy. Therefore, secondary fluorescence emission after two-photon excitation is indistinguishable from that observed in single-photon experiments. A fluorophore, such as rhodamine, will emit the same broad wavelength range of secondary fluorescence regardless of whether it was excited by a single or two-photon excitation event.
| Interactive Tutorial | |||||||||||
|
|||||||||||
Three-photon excitation is a related non-linear optical absorption event that can occur in a manner similar to two-photon excitation. The difference is that three photons must interact simultaneously with the fluorophore to illicit a transition to the excited singlet state. A benefit of three-photon excitation is that successful absorption requires only a tenfold greater concentration of photons than two-photon absorption, making this technique attractive for some experiments. Three-photon excitation can enhance z-axis resolution to an even greater degree than two-photon absorption. This is due to a smaller cross section for fluorophore excitation caused by the requirement for simultaneous interaction with three individual photons. In practice, a laser emitting infrared light with a wavelength distribution centered at 1050 nanometers is able to excite a fluorophore that absorbs in the ultraviolet region (approximately 350 nanometers, one-third of the excitation wavelength). The same laser can simultaneously excite another fluorophore at half the wavelength (525 nanometers), a useful combination in dual-labeled biological experiments.
By utilizing shorter near-infrared wavelengths (down to 720 nanometers), three-photon fluorescence can extend the useful fluorescence imaging range into the deep ultraviolet. Laser wavelengths in the 900 to 700 nanometer range will excite fluorophores that absorb in the 240 to 300 nanometer region, which is virtually inaccessible using conventional microscope optics. The glass used in manufacture of fluorescence objectives has very low transmission for wavelengths below 300 nanometers, but longer wavelength infrared laser radiation can easily pass through to produce three-photon excitation.
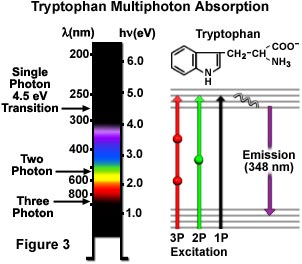
Single, dual, and triple photon excitations of a common aromatic amino acid, tryptophan, are schematically illustrated in Figure 3. A 4.5 electron volt single photon electronic transition excites tryptophan at 280 nanometers with the subsequent emission of secondary fluorescence at 348 nanometers in the ultraviolet region. Excitation by the two-photon mechanism is accomplished with greenish-yellow light centered at 580 nanometers, while three-photon excitation occurs when the amino acid is illuminated with 840-nanometer radiation in the near-infrared region. The transitions are presented in a Jablonski diagram (Figure 3), where the virtual state is represented by a sphere for two-photon excitation and by two spheres for three-photon excitation. Tryptophan has much stronger fluorescence with a higher quantum yield than the other aromatic amino acids, and is present only in small quantities in most proteins. These attributes should make multiphoton microscopy an excellent tool for investigations using autofluorescence of tryptophan residues. Even higher order nonlinear phenomena are possible, including four-photon excitation, but these have yet to be applied to biological research.
Two-Photon Fluorescence Microscopy
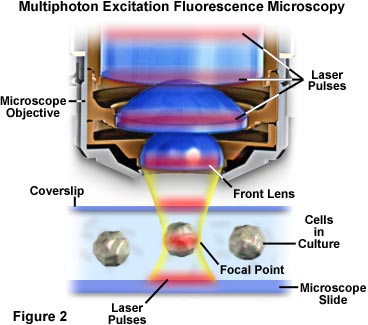
The localization of excitation to the region immediately surrounding the focal point in multiphoton microscopy occurs because it is here that the photon density is highest. This advantage arises from the basic physical principle that two-photon absorption by a fluorophore is a function of the square of the excitation intensity. When photons from a pulsed laser source are focused by a high numerical aperture objective, they become more crowded, thus increasing the probability that two or more will interact simultaneously with a single fluorophore. Concentration of photons at the microscope focal point is so critical to multiphoton absorption that this is the only region where appreciable excitation occurs. The concept is presented in Figures 2 and 4, which illustrate multiphoton excitation on a macroscopic and microscopic level, respectively. Figure 2 depicts an exaggerated view of a microscope objective in position to image cultured cells on a microscope slide and coverslip. Red laser pulses traverse the longitudinal axis of the objective and are focused and concentrated onto the cell in the central portion of the figure.
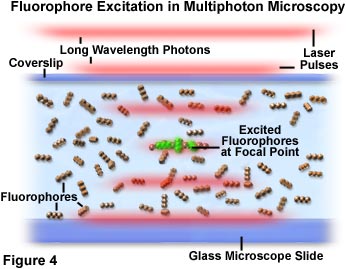
In Figure 4, photon crowding and interaction with fluorophores is demonstrated at the microscope focal point. As pulses of red laser light pass through the specimen containing fluorophores (represented as a linear triplet of spheres), the probability of excitation increases as the pulses reach the focal point of the objective. Individual photons are represented as an aggregate segregated into diffuse red lines that define the boundaries of laser pulses. A small group of fluorophore molecules positioned at the center of the focal region in Figure 4 have been excited by simultaneous absorption of two photons and are exhibiting green secondary fluorescence. There is nearly a zero probability that chromophores outside the focal plane will absorb two photons, because the photon density is not high enough in this region.
The phenomena of two-photon excitation is possible not only because of the spatial proximity of fluorophores at the microscope focal point, but also because of the temporal overlap of photons contained in sequential laser pulses. As mentioned above, the excitation energy in two-photon absorption occurs in proportion to the square of the photon intensity produced by the laser source. Pulsed laser beam intensity drops as the square of the distance from the focal plane, so the excitation probability of a fluorophore anywhere near the focal region decreases as the fourth power of the fluorophore's distance from the focal plane. The dimensions of the pulsed laser illumination cone are determined by the objective numerical aperture. Thus, the beam intensity decrease away from the focal point is proportional to the diameter of the excitation light cone squared. As the illumination cone expands above and below the focal point, fluorophore excitation probabilities decrease as the fourth power of the cone diameter. For this reason, fluorophore excitation is confined to the immediate region surrounding the focal point, which represents only a very thin optical section of the entire specimen.
| Interactive Tutorial | |||||||||||
|
|||||||||||
Laser pulse durations, which typically range from approximately 100 femtoseconds to 1 picosecond (10 e(-13) to 10 e(-12) second), are considered ultrashort in macroscopic terms. However, in terms of the time scale for photon absorption events (approximately one thousandth of a femtosecond) the pulses are actually quite long in duration. This limits fluorophore saturation and allows the molecules sufficient time to return to the ground state between pulses before another round of excitation occurs. Pulse repetition rates range from around 80 to 120 megahertz (MHz), which provides high instantaneous peak power for excitation, followed by a dwell time averaging 10 nanoseconds. Because the fluorescence lifetime of a typical fluorophore lasts only a couple of nanoseconds, the population of excited molecules has plenty of time to relax between pulses. The relatively short pulse duty cycle (the pulse duration time divided by the time between pulses) limits the average input laser power to less than 10 milliwatts, a value only slightly greater that that routinely employed for laser scanning confocal microscopy.
Limitation of two-photon excitation to the region near the focal plane provides a significant advantage for multiphoton over confocal microscopy. Fluorescence is excited throughout the specimen in confocal microscopy, but secondary fluorescence collected by the detector is restricted to the objective focal plane by the confocal pinhole. This serves to reduce the amount of background noise or fluorescence from other focal planes that add background noise to the data. In contrast, multiphoton microscopy generates fluorescence excitation (and subsequently, fluorescence emission) only at the focal plane, eliminating both the background signal and the necessity of a confocal pinhole. The dramatic difference between excitation modes in confocal and multiphoton microscopy is illustrated in Figure 5, which reviews photobleaching profiles for each technique.
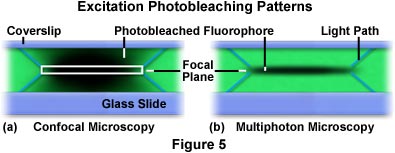
Presented in Figure 5 are the x-z photobleaching patterns that occur from repeated scanning of a single x-y plane in a formvar polymeric film stained with the fluorophore rhodamine (green stain). On the left (Figure 5(a)), is the profile generated by scanning the stained film with a confocal microscope. The white rectangle in the center of the scan represents the focal plane that is passed through the pinhole and imaged by the detector. Diagonal blue lines projecting from the upper and lower corners of the rectangle represent the light path taken by the excitation light beam through the film. As the beam raster scans the film, the fluorescent dye is excited and emits secondary fluorescence. Eventually, photobleaching occurs, which is represented by the dark areas in the focal region. In the film scanned by the confocal microscope (Figure 5(a)), the integrated excitation is nearly equal throughout the excitation path, both above and below the focal plane. Conversely, the x-z repetitive scan excitation profile generated by the multiphoton microscope limits excitation and photobleaching to the focal plane (Figure 5(b)). Similar to the case for Figure 5(a), the diagonal blue lines emanating from the focal plane delineate the path taken by the excitation light to reach the focal plane.
A number of advantages arise from the localized excitation afforded by multiphoton microscopy. Perhaps the most significant is the high degree of three-dimensional resolution that can be achieved with the technique, which is identical to that obtained with an ideal confocal microscope. Also, the lack of absorption from fluorophores positioned outside the focal plane allows more of the excitation light to penetrate through the specimen and reach the plane of focus. The result is a dramatically increased ability of the focused beam to penetrate deep within the specimen, frequently to a depth that can range between two and three times that observed with confocal microscopy.
As was discussed previously, the probability of multiphoton absorption outside the focal region drops as the fourth power of the distance along the optical axis (the z-direction). When a uniform distribution of fluorophores is subjected to multiphoton excitation with a high numerical aperture objective (1.4), approximately 80 percent of the absorption occurs in a tightly defined space termed the focal volume. The dimensions of this volume are dependent upon objective numerical aperture, but for a typical large aperture fluorescence objective at near-infrared wavelengths, this area is defined by an ellipsoid having a lateral dimension of 0.3 microns in diameter and an axial length of 1 micron.
| Interactive Tutorial | |||||||||||
|
|||||||||||
The significant reduction in the amount of photobleaching (and associated photodamage to cells and tissues) illustrated in Figure 5(b) for multiphoton microscopy is substantially less than occurs with confocal microscopy. Photobleaching and photodamage are two of the most important limitations of fluorescence microscopy in the study of living cells, tissues, and other organisms. Excitation of a fluorophore causes promotion of a ground state electron to an excited singlet energy state. During vibrational relaxation from the excited state, there is a probability that intersystem crossing will occur to a triplet state instead of the typical decay back to the singlet ground state. Triplet states are extremely reactive and relatively long-lived, which allows fluorophores in this condition time to react with living cells or to undergo molecular degeneration or rearrangement to a non-fluorescent species. In addition, excited fluorophores in a triplet state can generate singlet oxygen, which will react with a wide variety of functional groups on neighboring biomolecules. Excitation light must penetrate the specimen on all focal planes on the way to the focal point, and most of this light continues to propagate a considerable distance past the focal region. Thus, a population of fluorophores excited throughout the beam path, as is the case in widefield and confocal microscopy, will undergo a considerable amount of photobleaching and produce cell and tissue damage that can be avoided with the multiphoton technique.
Although the exact mechanisms of cell damage induced by exposure to light are poorly understood, it has been established that decreasing photodamage will dramatically extend the viability of biological samples investigated with fluorescence microscopy. Exposure to long-wavelength visible and near-infrared light alone does not appear to affect cell viability, so it is likely that a majority of the damage associated with multiphoton microscopy arises from excitation and is confined to the focal plane.
Detectors For Multiphoton Microscopy
In multiphoton microscopy, photons emitted through secondary fluorescence originate almost exclusively from the objective focal plane, eliminating the requirement for descanned detection and permitting more flexible detection geometries. This increased versatility can lead to a considerable improvement in fluorescence detection efficiency compared to confocal microscopy. In a system with descanned detection, light collected by the objective is reflected from the surface of a series of scanning mirrors before passing through a pinhole to the detector. While increasing resolution of the image, the confocal pinhole produces a large decrease in detection efficiency and necessitates longer exposure of the specimen to incident illumination, increasing the probability of photodamage and photobleaching.
In some cases it may be desirable to apply modified confocal detection techniques to multiphoton imaging (Figure 7). Invading room light can be excluded through the use of a large confocal pinhole, which can produce a slight increase in lateral resolution at the cost of signal collection efficiency. The pinhole also enables the utilization of detectors with small entrance apertures, such as avalanche photodiodes or spectrometers. Utilization of a pinhole for multiphoton imaging should be carefully scrutinized before being implemented. Much of the light emitted by the specimen that can contribute image formation will be blocked by the pinhole. This includes both light that is scattered from locations within the focal plane and light originating from the focus region boundaries. For the same reasons, there will also be a reduction in the amount of fluorescence collected from deep within the specimen. In fact, when confocal pinhole apertures are employed, signal falloff at increasing specimen depths will be similar for multiphoton and confocal imaging.
Several detection motifs are presented in Figure 7, which illustrates the collection of fluorescence information using photomultiplier tubes (PMTs), a CCD image array detector, and non-optical detection. Photomultipliers are the detectors of choice in multiphoton fluorescence microscopy, because shorter emission wavelengths (ultraviolet and low-wavelength visible) can be captured efficiently with these devices. The main distinction between the various scalar photomultiplier detectors in Figure 7 is whether emitted fluorescence is passed back through the scanning mirrors (descanned detection) or relayed through a transfer lens to a detector (PMT) placed in a plane conjugate to the objective rear aperture. A third photomultiplier (labeled the external detector) is shown positioned to the lower right of the specimen, and is designed to capture fluorescence directly from the specimen without passing through any portion of the microscope optical train.
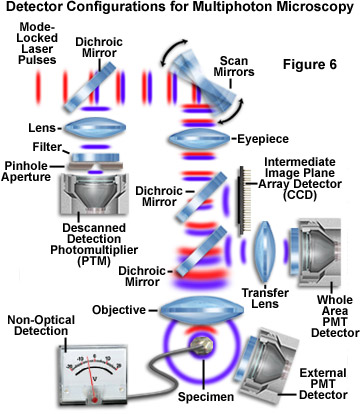
Because of the fact that resolution is defined by the multiphoton excitation process, light emitted by excited fluorophores can be collected without using the objective. In fact, a high numerical aperture condenser will adequately serve the purpose of accumulating secondary fluorescence emission for the detector. In some cases, a photodetector is placed above (or below, depending upon microscope configuration) the specimen in an area removed from the microscope optics (Figure 7). This detection scheme enables the utilization of short wavelength emission that might be hampered or precluded by low transmission through objective lenses. Another strategy is to collect emission directly from the objective with a detector (such as the whole area photomultiplier, as discussed above) placed near a dichroic mirror that reflects light from the rear aperture through a transfer lens. A shorter emission pathway facilitates the number of photons collected, especially when imaging through ten to twenty microns of tissue bathed in saline. Although the latter method can be employed to maximize the fluorescence detection efficiency, it is often vulnerable to contamination by ambient room light.
Photodiode arrays, such as a charge-coupled device (CCD) or complementary metal-oxide semiconductor (CMOS) detector, can also be utilized to collect fluorescence information in multiphoton experiments (Figure 7). In this configuration, light emitted by the specimen is again reflected from a dichroic mirror and imaged directly onto the photodiode array surface, which is placed in a plane conjugate to the intermediate image plane. An improvement in lateral resolution can often be obtained with this technique because resolution is determined by the emission wavelength, which is shorter than the excitation wavelength.
Several non-optical detection schemes have the potential to be applied to multiphoton microscopy due to the high degree of spatial localization during excitation. Among these are opto-acoustic detection, used to measure small amounts of absorption, and scanning photochemical detection, which generates images of receptor distributions from ionic current in voltage-clamped cells.
Resolution in Multiphoton Microscopy
Resolution in multiphoton microscopy does not exceed that achieved with confocal microscopy and, in fact, the utilization of longer wavelengths (red to near-infrared; 700 to 1200 nanometers) results in a larger point spread function for multiphoton excitation. This translates into a slight reduction of both lateral and axial resolution. For example, with an excitation wavelength of 700 nanometers and a 1.3 numerical aperture objective, the observed lateral resolution is approximately 0.2 micrometers with a corresponding axial resolution of 0.6 micrometers. When coupled to the Stoke's shift size, these values can range up to 30 percent larger than the resolution observed with conventional confocal microscopy under identical conditions. In practice, confocal resolution can be degraded by the finite pinhole aperture, chromatic aberration, and imperfect alignment of the optical system, all of which serve to reduce resolution differences between confocal and multiphoton microscopy. From this discussion, it becomes apparent that if structures are not adequately resolved with a confocal microscopy, they will not fare any better (and may be worse) when imaged with multiphoton excitation.
When gathering digital images or counting photons with three-dimensional spatial resolution, it is essential to distinguish between fluorescence emission occurring within the focal volume from that originating in the background. Differentiation between the two signals can be accomplished instrumentally (with confocal or multiphoton instrumentation) or by deconvolution of a three-dimensional data set. The ability to distinguish between fluorescence emission from the focal plane and background fluorescence is defined by the signal-to-background (S/B) ratio, where S is the number or intensity of photons collected from the focal plane and B represents the photons originating from the background (out-of-focus planes). In confocal scanning microscopy, high S/B ratios are generated by rejection of background signal by the confocal pinhole. However, in multiphoton excitation, S/B ratios are inherently large because there is very little excitation outside the focal plane. Resolution calculations between multiphoton and confocal techniques can be compared by considering an infinitely small pinhole when performing the confocal calculations. For both techniques, the signal-to-background ratio is typically several orders of magnitude larger than for classical widefield fluorescence microscopy.
Another point to consider is that multiphoton excitation enables the utilization of fluorophores with absorption transitions in the low wavelength ultraviolet region. Because confocal microscopy is limited in its ability to excite fluorophores below about 340 nanometers, investigators tend to utilize probes having much longer wavelengths, with correspondingly lower resolutions. In critical situations, resolution in multiphoton microscopy can be enhanced through the restriction of imaging wavelengths via a confocal pinhole, and also by utilizing a spatially resolved detection system such as a CCD photodiode array placed in a scanned image plane.
Excitation Characteristics of Fluorophores
Fluorophores employed in multiphoton experiments should be subjected to the same scrutiny as those intended for single-photon investigations. The probes should have large absorption cross-sections at convenient wavelengths, high quantum yields, a low photobleaching rate, and the lowest possible degree of chemical and photochemical toxicity. The fluorophores should also be able to withstand high intensity illumination from the laser source without significant degradation. In most cases, investigators have utilized the same common fluorophores for two-photon experiments that are widely applied as markers in widefield and confocal fluorescence microscopy.
The excitation spectrum of common fluorophores is a function of the excitation mode and the wavelength of incident photons. Because of this dependency, the two-photon absorption spectrum can (and often does) differ dramatically from the corresponding single-photon spectrum. Experimentally, a majority of the fluorophores that have been examined are capable of absorbing two-photon excitation at twice the wavelength of their one-photon maximum absorption. In spite of this, there is no fundamental basis for quantitatively predicting the two-photon excitation spectrum of a complex fluorophore simply by examining the single-photon cross section. Significant differences often exist between the single and two-photon excitation spectra for highly conjugated non-symmetrical molecules, which are often exploited in molecular spectroscopy to provide information about the structure of excited states. A good example is the aromatic amino acid derivatives tyrosine and phenylalanine, whose complex two-photon cross-sections are quite different from that displayed by single-photon excitation. In contrast, the two-photon spectrum for tryptophan (Figure 2) is very similar to the profile displayed for single-photon excitation.
For quantitative three-dimensional reconstruction and deconvolution experiments, the two-photon absorption spectra of fluorophores should be measured to ensure that excitation wavelengths are centered near peaks in the absorption bands. Although two-photon cross-sections can be calculated, the process is complex at best. Direct experimental measurement of the absorption spectra is the preferred method, however these experiments are difficult due to the small amount of incident power absorbed versus intensity fluctuations in the light source. Thermal lensing and acousto-optical techniques have been employed to determine absorption cross-sections, but perhaps a simpler method is to examine photon emission from fluorophores with known quantum yield. When designing new two-photon experiments, a range of fluorophores should be examined having absorption peaks near the one-half value of the intended excitation wavelength.
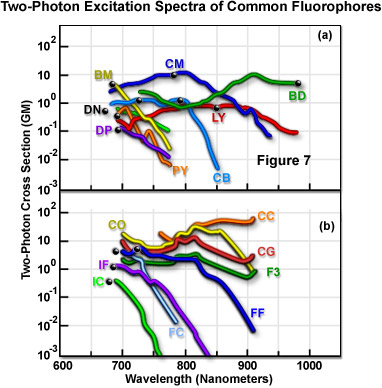
Figure 7 presents the characteristics of measured two-photon fluorescence excitation spectra for a number of common fluorophores. The data in Figure 7 represents two-photon action cross-sections, which is derived by taking the product of the fluorescence emission quantum efficiency and the two-photon absorption cross-section. Spectra were recorded utilizing linearly polarized light emitted by a mode-locked Ti:sapphire laser. In each spectrum, the black dot represents twice the wavelength of the fluorophore's single-photon absorption maximum. Table 1 is a key to the two-letter name codes presented beside each spectrum in Figure 7. The curves represent the spectral cross-sections of fluorophore two-photon excitation.
Two-Photon Fluorescence Excitation Spectra of Fluorophores
|
||||||||||||||||||||||||||||||||||||
Table 1
Cross-section measurements indicate a trend in which the excitation peak for two-photon absorption is very similar or blue-shifted with respect to the single-photon profile (Figure 7). The shorter average wavelengths may be advantageous in coupling fluorophore excitation to the available wavelength range of mode-locked lasers. Another consistent aspect of two-photon absorption spectra is that they are usually much broader than their single-photon counterparts. This eases experimental constraints by increasing the range of wavelengths that are suitable for excitation and enhances the ability to simultaneously excite two fluorophores that have overlapping two-photon cross-sections, but widely separated single-photon spectra. Measurements of three-photon cross-sections indicate that they are, in general, very similar to the corresponding single-photon spectra.
Although absorption spectra often differ for single and two-photon excitation, other fluorescence properties such as lifetime, emission wavelengths, and the rate of intersystem crossing do not appear to be affected. This similarity indicates that the same fluorescence excited states are reached by either linear or nonlinear absorption, and that once the fluorophore has been excited, it will behave the same regardless of the excitation mode. These tenants also hold for three-photon excitation, allowing investigators to utilize well-established ratiometric and spectroscopic methods in a majority of multiphoton experiments.
Photo and Heat Damage in Multiphoton Excitation
All forms of fluorescence microscopy suffer from photodamage to living cells, the degree of which is dependent upon the excitation wavelength, length of exposure, and the chemical nature of fluorophores utilized as cellular probes. The damage induced by excitation illumination can be segregated into two categories: heat damage and degradation due to chemical reactions. Photochemical side effects caused by biochemical reactions, as a result of fluorophore excitation, are not well understood and vary widely among cell and tissue types. Heat damage, on the other hand, arises principally from two mechanisms that occur because of single-photon absorption by water and by two-photon absorption from fluorophores in the focal region.
In most cells studied (particularly mammalian cells), there is almost no absorption of long wavelength near-infrared excitation radiation by intrinsic fluorophores utilized in multiphoton fluorescence. However, intracellular and intercellular water surrounding cells and tissues can absorb significant amounts of infrared and near-infrared illumination, producing excess heat that is potentially damaging to the viability of biological specimens. On the other hand, when the aqueous biological environment is illuminated with the shorter visible and ultraviolet wavelengths utilized in confocal and widefield fluorescence microscopy, a significant amount of heat is not absorbed by the surrounding water.
Heating due to single-photon absorption by water occurs all along the beam path, both above and below the focal plane. Under controlled average multiphoton conditions, the induced temperature increase has been calculated to range between 0.065 and 1.1 degrees Centigrade at 700 and 1000 nanometers, respectively. These calculations are in agreement with heat measurements conducted at 1064 nanometers with optical tweezer laser excitation. In situations where the exciting light beam is held stationary, greater heating can occur, rising rapidly in a logarithmic relationship with time. Heating due to fluorophore absorption is highly localized to the focal region in multi-photon excitation experiments. Subsequent heat release occurs uniformly within a spherically symmetrical region surrounding the focal volume, and produces no significant amount of heat, even at high fluorophore concentrations.
Conclusions
Multiphoton fluorescence microscopy is becoming one of the methods of choice for dynamic imaging of living cells and tissues. The technique is particularly useful in biological systems where ultraviolet excitation would not otherwise be possible due the light transmission characteristics of optical systems. In addition, side effects such as photobleaching and photodamage are minimized in multiphoton excitation, and occur only in the immediate region surrounding the focal volume. Although the prediction and measurements of two-photon absorption profiles from common fluorophores are slowly being accomplished, a significant amount of work in this area is left to be completed. Design of new fluorophores specific for multiphoton excitation remains in the embryonic stages, but some progress along this avenue should be expected over the next few years.
Phototoxicity in cells is a poorly understood phenomenon, but does occur to a large degree in most forms of fluorescence microscopy. The lower quantum energy and lower intrinsic absorption of longer wavelengths utilized in multiphoton microscopy serve to reduce the deleterious effects of light on living cells and tissues, opening the door to new investigations of cellular dynamics. A major impediment to research in multiphoton microscopy is the high cost of equipment, in particular the necessary mode-locked pulsed laser systems that are required for two-photon and three-photon excitation. The two main types of ultrafast laser systems in general use are Ti:sapphire and Nd:YLF lasers, which although are very expensive, do not require water cooling nor have excessive electrical power demands. The wavelength tunability of the Ti:sapphire pulsed laser (700 to 1100 nanometers) renders it far more versatile than the single-wavelength Nd:YLF laser (1047 nanometers), but the convenience of tunability precludes total hands-off operation. As newer, cheaper, and user-friendly lasers are introduced into a competitive market, more commercial multiphoton microscope systems will be introduced at a cheaper price. This availability, coupled to the fact that there are no physical limitations to the implementation of multiphoton fluorescence, will encourage the widespread application of this technique throughout the biological sciences.
Contributing Authors
David W. Piston - Department of Molecular Physiology and Biophysics, Vanderbilt University, 702 Light Hall, Nashville, Tennessee, 37212.
Michael W. Davidson - National High Magnetic Field Laboratory, 1800 East Paul Dirac Dr., The Florida State University, Tallahassee, Florida, 32310.
BACK TO MULTIPHOTON MICROSCOPY