
Fluorescence Microscopy
Anatomy of the Fluorescence Microscope
In contrast to other modes of optical microscopy that are based on macroscopic specimen features, such as phase gradients, light absorption, and birefringence, fluorescence microscopy is capable of imaging the distribution of a single molecular species based solely on the properties of fluorescence emission. Thus, using fluorescence microscopy, the precise location of intracellular components labeled with specific fluorophores can be monitored, as well as their associated diffusion coefficients, transport characteristics, and interactions with other biomolecules. In addition, the dramatic response in fluorescence to localized environmental variables enables the investigation of pH, viscosity, refractive index, ionic concentrations, membrane potential, and solvent polarity in living cells and tissues.
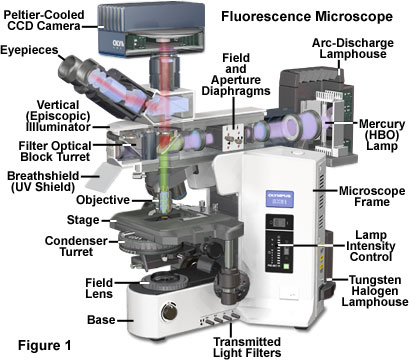
The earliest fluorescence microscope configurations featured a classical brightfield or darkfield diascopic (transmitted light) optical train that focused excitation light passed through a filter onto the specimen plane. Fluorescence emission was gathered by the objective, along with a significant amount of the excitation illumination, and projected through a second filter into the eyepiece diaphragm to form the intermediate image. Because the intensity of excitation light is usually several orders of magnitude greater than fluorescence emission, the specimen view in these early transmitted light microscopes was often very low in contrast and superimposed on a background flooded with scattered excitation illumination. Using a high numerical aperture oil-immersion darkfield condenser to illuminate the specimen at highly oblique azimuths helped to eliminate most of the background noise, but did not provide adequate illumination for any but the lowest numerical aperture objectives. The resulting images suffered from poor resolution and very low brightness levels.
Fluorescence microscopy with incident (reflected light or episcopic) illumination was first developed in the late 1920s to observe fluorescence emission in opaque metallurgical specimens. Like their brightfield counterparts, these reflected light fluorescence instruments employed half-mirror beamsplitters that are restricted to an overall efficiency of approximately 25 percent (after losing 50 percent of the illumination intensity upon each pass through the mirror). However, reflected light fluorescence microscopes enjoyed the advantage of having a high numerical aperture objective acting as the condenser, and were able to produce images with a significantly greater level of brightness than transmitted light microscopes operating at similar numerical apertures. In addition, the stray excitation light reflecting back into the objective (at high numerical aperture) was reduced to only a few percent. The dual role of the objective in reflected light microscopy also significantly eases the task of alignment. Merely focusing the objective onto the specimen establishes both the illumination field and the field of view such that incident excitation light and the observed fluorescent emission follow identical pathways through the microscope optical train.
The move to reflected light fluorescence (at least for metallography) was bolstered by technical advances in illumination sources when compact mercury-vapor and xenon arc-discharge lamps were developed in the mid-1930s. During this period, colored glass and gelatin filters were also becoming more sophisticated, which enabled the application of fluorochromes excited by blue and green visible light using tungsten-halogen lamps. Anti-reflective coatings and improved glass formulations led to major improvements in objective design during the 1940s, but the most fundamental contribution to the development of incident light fluorescence microscopy was the introduction of dichromatic beamsplitters (also termed dichroic mirrors) by Russian optical scientist Eugeny Brumberg in 1948. This innovation overcame the light loss problems inherent with the use of ordinary half-mirrors in reflected light microscopy. Reflected light fluorescence microscopes were first commercialized on a broad scale by Johan S. Ploem in the late 1960s, who was instrumental in developing the Wild-Leitz Ploem Opak, containing multiple optical blocks that were interchangeable and housed various combinations of filters for fluorescence microscopy.
Basic Strategy of Epi-Fluorescence Microscopy
Reflected light fluorescence microscopy is overwhelmingly the current method of choice for widefield investigations with non-coherent light sources, as well as those conducted with laser scanning confocal and multiphoton instruments. This popular mode of fluorescence microscopy is also known as incident light fluorescence, episcopic fluorescence, or simply epi-fluorescence. A typical modern reflected light fluorescence microscope, also equipped for transmitted light observation in a variety of contrast-enhancing modes, is presented in Figure 1. The microscope contains a trinocular observation head that is coupled to a charge-coupled device (CCD) camera system, and has two illumination sources, one for transmitted light and the other for episcopic observations (tungsten-halogen and mercury arc-discharge, respectively). A microscope of this design can combine or alternate reflected light fluorescence with transmitted light phase contrast, differential interference contrast (DIC), polarized light, or Hoffman modulation contrast observation.
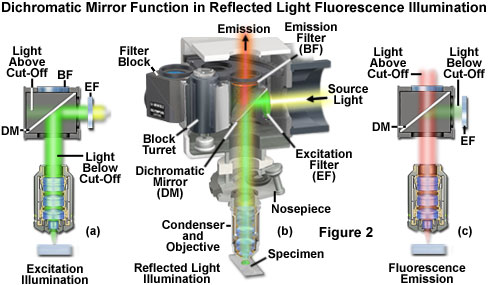
The essential feature of any fluorescence microscope is to provide a mechanism for excitation of the specimen with selectively filtered illumination followed by isolation of the much weaker fluorescence emission using a second filter to enable image formation on a dark background with maximum sensitivity. Localized probe concentration in biological specimens is so low in many experiments that only a small fraction of the excitation light is absorbed by the fluorescent species. Furthermore, of those fluorophores that are able to absorb a quantity of illumination, the percentage that will emit secondary fluorescence is even lower. The resulting fluorescence emission brightness level will range between three and six orders of magnitude less than that of the illumination. Thus, the fundamental problem in fluorescence microscopy is to produce high-efficiency illumination of the specimen, while simultaneously capturing weak fluorescence emission that is effectively separated from the much more intense illumination band. These conditions are satisfied in modern fluorescence instruments by a combination of filters that coordinate excitation and emission requirements based on the action and properties of the dichromatic beamsplitter.
The principles behind dichromatic beamsplitter (mirror) function in reflected light fluorescence microscopy are outlined in Figure 2 for a hypothetical specimen containing a fluorophore that is excited in the green region (550 nanometers) and fluoresces in the red (620 to 660 nanometers) wavelengths of the visible light spectrum. A high-intensity light source outputs a wide spectrum of excitation wavelengths at high flux density (usually covering most of the ultraviolet and the entire visible spectrum), which travels through the illuminator and first encounters a filter that selects the proper wavelength band for excitation (labeled EF; see Figure 2(a)). In this case, the filter passes light having wavelengths between 510 and 560 nanometers with high efficiency, but also allows other wavelengths to pass at a much lesser extent. The excitation light next reaches the dichromatic mirror (DM in Figure 2) and is reflected into the objective rear aperture to form a cone of illumination that bathes the specimen. The dichromatic mirror is positioned in the light path at a 45-degree angle and designed to selectively reflect wavelengths between 490 and 565 nanometers (as illustrated in Figure 3), while simultaneously transmitting both shorter and longer wavelengths.
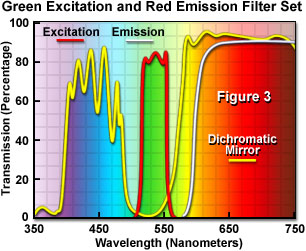
Presented in Figure 3 are the transmission profiles for the filter combination used to separate excitation illumination from fluorescence emission in Figure 2. The excitation filter spectrum (red curve) exhibits a high level of transmission (approximately 80 percent) between 510 and 560 nanometers with a center wavelength (CWL) of 535 nanometers. The dichromatic mirror (yellow curve) reflects wavelengths in the region of the excitation spectrum, while passing higher and lower wavelengths with relatively high efficiency. Note that zero percent transmission on the dichromatic mirror curve corresponds to 100 percent reflection. The pronounced dip in the transmission profile between 490 and 570 nanometers, which represents a peak in reflectance, serves to reflect the band of wavelengths passing from the excitation filter at a 90-degree angle and onto the specimen. The final component in the optical train, an emission or barrier filter (white curve), transmits wavelengths higher than 590 nanometers, which corresponds to visible light having yellow, orange, and red colors. Boundaries between transmitted and reflected wavelength bands of the various superimposed spectra are designed to be as steep as possible to assure nearly complete separation of the reflected and transmitted wavelengths. A pattern of sinusoidally rising and falling spikes appearing in the dichromatic mirror spectrum is a common effect of the thin-film deposition process known as ringing. The performance of this filter combination is remarkable and is a clear demonstration of the rapid advances being achieved in thin film interference filter technology.
Because only a narrow bandwidth of light is reflected by the dichromatic mirror, illumination wavelengths shorter than 490 nanometers and longer than 565 nanometers that manage to pass through the excitation filter are also transmitted through the dichromatic mirror, illustrated as the light above the cut-off in Figure 2(a). Note that the reflection of excitation light is not 100-percent efficient, and thus, a small amount of green light passes through the dichromatic mirror without being reflected. In addition, not all of the light having wavelengths above 565 or below 490 nanometers is transmitted through the mirror. A small percentage of this light is reflected by the mirror through the objective and onto the specimen. Light transmitted from the excitation filter through the dichromatic mirror (light above the cut-off in Figure 2(a)) is partially absorbed by the flat black coating on the interior of the filter block, but some reflects from the surface and passes through the barrier filter at an oblique angle, contributing to the fluorescence background noise.
Fluorescence emission by the specimen (primarily red wavelengths), which results from the green light excitation, is gathered by the objective and passes through the dichromatic mirror and barrier filter (light above cut-off in Figure 2(c)). The barrier filter (labeled BF in Figure 2) is specifically designed to allow only light of wavelengths greater than 590 nanometers to reach the microscope eyepieces and/or detector. In serving this duty, the barrier filter effectively prevents excitation light wavelengths reflecting from the specimen (and successfully traversing the dichromatic mirror) from reaching the detector. However, a majority of the excitation wavelengths returning from the specimen are reflected towards the excitation filter and illuminator by the dichromatic mirror (the light below cut-off in Figure 2(c)). The net effect of the filter configuration illustrated in Figures 2 and 3 is to separate the excitation light, which is substantially higher in intensity, from the much weaker fluorescence emission. In all cases, as shown in the figures, the cut-off levels of the filters involved in fluorescence microscopy are not absolute, but enable some light outside the wavelength range to bleed through. The entire sequence of events is graphically presented in Figure 2(b), which illustrates the optical pathways and strategic filter arrangement necessary for efficient reflected light fluorescence microscopy.
The Vertical Fluorescence Illuminator
At the heart of the modern fluorescence microscope is the universal reflected light vertical illuminator, which is interposed between the observation viewing tubes and the nosepiece carrying the objectives, as illustrated in Figures 1 and 4. The illuminator is designed to direct light generated by a high-intensity source onto the specimen by first passing the light through the microscope objective on the way toward the specimen and then using that same objective to capture the light being emitted by the specimen. This type of illumination strategy has several advantages. The microscope objective, which acts first by serving as a well-corrected condenser, next gathers image-forming fluorescence emission for transmission to the eyepieces or camera detection system. As such, the objective is always in correct alignment relative to each of these functions. Furthermore, most of the excitation light that is scattered or reflected by the specimen (over a 360-degree angle) travels away from the objective front lens element, rather than being projected directly into the glass, as is the case in transmitted fluorescence illumination. This effect is referred to as front-face illumination and is particularly useful with thick specimens. Finally, the specimen area being illuminated is restricted to the same area that is being observed, and both illumination and light collection can utilize the full numerical aperture of the objective.
At the far end of the vertical illuminator is the lamphouse (see Figure 4), which contains a high-intensity arc-discharge or filament-based incandescent light source. The most popular illumination source is a high-pressure 100-watt mercury arc lamp (HBO, often termed a burner), but xenon and metal halide arc lamps, lasers, and tungsten-halide incandescent lamps can serve the purpose as well. Light emitted by the source is focused by the collector lens system and travels along the interior of the illuminator parallel to the tabletop and perpendicular to the microscope optical axis. The illuminator design illustrated in Figure 4 contains a multiple-component collector lens system, but aspherical lens elements are also being implemented to improve correction for chromatic aberration in the near-infrared and ultraviolet regions. A heat filter designed to remove or suppress infrared wavelengths is placed either inside the lamphouse itself or adjacent to the lamphouse mount at the rear of the vertical illuminator. In addition, some microscopes contain a dual or multi-band excitation balancer system (see Figures 4 and 8) near the lamphouse to selectively filter wavelengths that will not be needed for excitation in order to fine-tune the performance of the fluorescence filter set contained at the other end of the illuminator.
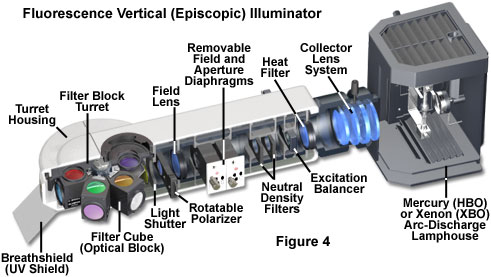
Also positioned near the lamphouse in the vertical illuminator are a set of neutral density filters that can be employed to adjust the overall intensity of light passing through the system and reduce fluorescence fading or photobleaching. These filters are usually mounted on slider frames that enable them to be quickly inserted and removed from the light path while viewing the specimen fluorescence. In order to establish Köhler illumination, the vertical illuminator contains a centerable aperture and field diaphragm combination, both of which have variable iris apertures that dictate the field size and illumination intensity. Centering of the aperture and field diaphragms and adjustment of the iris size is accomplished with several pairs of knobs located on each exterior side of the vertical illuminator housing. In some designs, a slider knob is provided that removes the entire diaphragm assembly from the light path to maximize the amount of light passing through the illuminator. Other microscopes contain a pinhole aperture of fixed size mounted on a slider that can be inserted into the light path to dramatically restrict the illumination field for specialized applications, such as fluorescence recovery after photobleaching (FRAP) investigations.
Following the field and aperture diaphragms in the vertical illuminator optical train is the field lens, which is necessary to spread the light and create a sufficient illumination field for establishing Köhler illumination. After the field lens, all modern vertical illuminators contain a light shutter (with manual on-off control) that blocks the intense excitation light from reaching the filter sets and specimen when fluorescence is not being observed or recorded. Shutters are a critical feature in fluorescence microscopy because continuous illumination of the specimen can dramatically reduce emission due to photobleaching, and high-intensity light is unhealthy for living cells. In addition, a shutter enables the arc-discharge lamp to remain active (alleviating a warm-up period) while specimens are being transiently examined with transmitted light. High-end fluorescence microscopes are often equipped with electronic shutters that provide rapid and remote control of illumination. An ultraviolet protection shield (labeled breathshield in Figure 4) is fitted onto the front of the illuminator housing to protect the operator from any inadvertent leakage of potentially dangerous short-wavelength ultraviolet radiation when the shutter is open. The shield serves double duty by also protecting the specimen from exhaled breath while under observation.
Several vertical illuminator designs provide a slot for rectangular polarizer frames that can be employed in fluorescence polarization investigations. In most cases, the polarizer is inserted into the illumination light path behind the field lens (away from a conjugate plane), but preceding the light shutter (see Figure 4). The polarizer can be either fixed into position with a pre-determined transmission azimuth, or mounted onto a gearset, allowing the transmission axis orientation to be varied through the use of a thumbwheel. Because the polarizer is mounted in a sliding frame, it can be easily removed from the light path when not in use. The accompanying analyzer (a second polarizer necessary for fluorescence anisotropy measurements) can be mounted in the vertical illuminator above the filter block turret, or in a specially designed intermediate tube for polarized light microscopy. The auxiliary tube often enables insertion of a graduated, rotating polarizer unit to ensure accurate positioning of the analyzer transmission azimuth with respect to the polarizer and microscope optical axis.
The last stage of the vertical illuminator contains a revolving turret or sliding bracket that houses optical blocks containing the fluorescence filter combinations. The filter blocks are rotated (or slid) into the optical train when required for imaging with the specific fluorescence filter combinations. Individual blocks contain a matched set of excitation and emission filters, as well as a dichromatic beamsplitter, all carefully positioned to maximize specimen illumination and capture of fluorescence emission at specific wavelengths. Optical block turrets and slider brackets can load between three and 6 individual blocks with one positioned in the light path, while the others are rotated or slid out of the way in temporary storage. These accessories enable rapid changeover between fluorescence filter sets when observing specimens labeled with two or more fluorescent probes. The use of transmitted illumination in a microscope equipped with a fluorescence illuminator is facilitated by dummy optical blocks that fit into a slot on the turret or slider bracket. The dummy blocks block excitation emission (when the shutter is opened), but do not contain any filters and allow light to pass unimpeded from the objective to the observation tubes.
In standard modular upright microscopes, the vertical illuminator is positioned between the microscope frame and the observation tubes (see Figure 1). Many manufacturers offer intermediate tubes that can be inserted between the illuminator and the eyepiece unit for polarizers, DIC prisms, or other accessories. Modern microscope frames are usually the result of computer-aided design efforts that result in significant vibration reduction and enhanced ergonomic features. Synthetic materials, such as metallic matrix ceramic and aluminum composites, dramatically improve the static and thermal rigidity of the frame, thus enabling the instrument to withstand the rigors of advanced fluorescence techniques that require an almost total elimination of vibration over the long periods of time necessary for imaging weak fluorescence emission.
Köhler Illumination in Fluorescence Microscopy
In a fluorescence vertical illuminator, the light source is positioned so that the filament or arc-discharge plasma ball is located near the principal focal point of the collector lens. In Köhler illumination, the lamp collector lens serves the function of a dramatically enlarged secondary light source to enhance overall illumination. One the primary requirements of Köhler illumination is that an image of the lamp filament or the arc must ultimately be projected onto the rear focal plane of the objective, which also doubles as the (often high numerical aperture) condenser during excitation in reflected light illumination. The light source should ideally fill the entire objective aperture to both maximize the intensity of radiation and to produce an evenly illuminated field. In some cases, a ground glass filter is placed into the vertical illuminator between the lamphouse and the neutral density filters in order to increase the uniformity of illumination, especially when employing arc lamps that tend to produce regions of excessive intensity (termed hot spots). However, because diffusion filters also reduce the level of illumination, they should be avoided whenever possible.
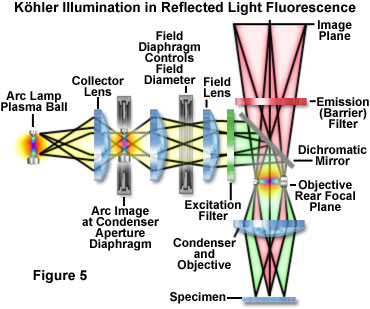
In reflected light Köhler illumination (illustrated schematically in Figure 5), an image of the light source is focused by the collector lens onto the aperture iris diaphragm located in the vertical illuminator. This diaphragm shares a conjugate plane with the rear aperture of the objective and the lamp arc or filament, and therefore, determines the illuminated field aperture size. Together, the light source, vertical illuminator aperture diaphragm, and objective rear focal plane (pupil) form the illumination set of conjugate planes. Unlike the situation in transmitted light microscopy, the aperture iris and light source are imaged onto the objective (acting as a condenser) rear aperture plane, rather than being physically located at this position. As an added benefit to this configuration, all obstructions (such as iris diaphragms) are removed from the light path when the objective is providing excitation illumination or forming an image from the gathered fluorescence emission. Opening or closing the aperture diaphragm is used to control stray light and regulate the intensity (numerical aperture) of illumination without altering the size of the illuminated field. In the image, adjustment of the aperture diaphragm affects brightness and contrast.
The image-forming or field set of conjugate planes in reflected light Köhler illumination consists of the field diaphragm, the specimen surface, and the intermediate image plane. Thus, when the field diaphragm is placed in focus at the specimen plane, the image of the light source is significantly removed from focus in order to provide a uniform field of illumination. The field diaphragm controls the size of the illuminated field without affecting the illumination intensity of the area being observed. In practice, the field diaphragm opening size should be as small as possible in order to increase image contrast and to reduce the degree of photobleaching in areas not being directly observed. Köhler illumination produces even illumination of the specimen field in spite of the uneven illumination intensity generated by most arc-discharge and filament-based light sources. When the microscope is properly configured, the rear focal plane of the objective is fully illuminated, providing a field that is uniformly bright from edge to edge. Köhler illumination, in the ideal case, bathes the specimen with a converging set of wavefronts, each arising from separate points on the light source imaged into the condenser aperture. In a properly configured fluorescence microscope, the result is optimum image contrast and resolution.
Light Sources and Lamphouses
For rigorous quantitative analysis in fluorescence microscopy, specimen illumination must be temporally and spatially constant over the entire viewfield. Instability over time generally reflects fluctuations of lamp radiance that occur as a result of electrical power supply output variations. In contrast, spatial disturbances, which occur most often in arc lamps, arise from a phenomenon known as flicker as the plasma ball migrates back and forth across the electrodes. Flicker is generally produced by power supply fluctuations, small variations in the electrode resistance, or mechanical vibrations. Filament-based light sources, such as the popular tungsten-halide lamp, are very stable when operated under constant direct current with a regulated power supply. In general, light sources should be chosen from their spectral content, filament size compared to the objective rear aperture area, spatial and temporal stability, and uniformity of field illumination. Careful attention must also be given to the intensity of the light source because the narrow band of wavelengths passed by the excitation filter comprises only a very small portion of the total illuminator output.
The primary consideration in choosing a light source for fluorescence microscopy is the ultraviolet and visible light spectral distribution in relation to the quantum yield and absorption of fluorochromes being investigated. In addition, the source must be compatible with the sensitivity of the detector used of capture images, whether it is the human eye, traditional film, a photomultiplier, intensified video tube, or a digital camera system. The choice also depends on the mode of illumination. Widefield fluorescence microscopy requirements are fulfilled with arc-discharge or tungsten-halogen sources, while confocal and multiphoton microscopy requires the adaptation of various laser systems. Tungsten and tungsten-halogen incandescent lamps have been used with limited success due to the fact that a majority of their emission occurs in the red and infrared regions of the light spectrum, whereas most fluorophores are excited by ultraviolet, blue, and green wavelengths. In addition, the light output from arc-discharge lamps ranges between 10 and 100 times brighter than the 12-volt quartz halogen lamps typically used for transmitted light illumination. The favorite source of illumination for widefield fluorescence microscopy is the mercury arc lamp, which is usually routinely included in base-model microscope configurations. In some cases, xenon and metal halide arc lamps are employed, but these are usually confined to specialized circumstances where the light spectrum and intensity profile matches specific fluorophore requirements.

The design of mercury and xenon lamps is similar, except for the physical dimensions and the gas enclosed in the bulb envelope. Mercury lamps contain two electrodes sealed under high pressure in a quartz glass bulb that also contains vaporized elemental mercury. When the power supply is energized, a series of high voltage pulses is sent to the electrodes, which ionizes a small portion of the mercury gas, igniting the lamp. After firing, the voltage is reduced and the ionized gas serves to carry current and generate the plasma ball that forms between the two electrodes. During lamp operation, the vaporization of mercury generates a tremendous amount of heat and pressure inside the glass bulb, ultimately producing the high intensity light. Mercury arc lamps produce light that, even though continuous across the ultraviolet and visible spectrum, is concentrated in discrete wavelengths of 365, 400, 440, 546, and 580 nanometers. Xenon lamps have a more uniform intensity profile across the entire spectrum, from the ultraviolet to the infrared. The choice of fluorochromes is paramount in determining the proper light source for fluorescence microscopy. Some fluorescent probes have excitation bands that coincide with the prominent mercury lines, while others benefit from the more evenly distributed illumination of a xenon lamp.
Proper alignment of arc lamps in fluorescence microscopy is critical in order to achieve Köhler illumination and to avoid bright and dark regions in the fluorescence image. As such, the quality of the lamphouse can often be judged by the stability of correct lamp alignment and by the efficiency of the adjustment knobs for maintaining the alignment. The lamp socket should be equipped with lamp centering screws to permit centering the arc image in the objective rear aperture, and the lamphouse should incorporate an infrared filter to block the very long wavelengths in the far red and infrared that generate a tremendous amount of heat. Several lamphouse designs have a built-in red suppression filter (for example, a Schott BG38), or a slot for such a filter, to eliminate a reddish background seen through the viewfield in some applications. In addition the lamphouse itself should not leak harmful ultraviolet wavelengths and, preferably, should incorporate a switch to automatically shut down the lamp if the housing is inadvertently opened during operation. Finally, the lamphouse should be sturdy enough to withstand a possible burner explosion during operation.
The wide diversity of fluorescence microscopy applications often call for a range of light sources to meet the demands of specific fluorophores and imaging conditions. In some cases, very low irradiation may be required in combination with an ultra-sensitive camera system, whereas for other investigations, strong laser excitation may be necessary in order to kill living cells or selectively bleach a fluorophore. Wavelength requirements often span the entire visible region of the spectrum, as well as portions of the ultraviolet and infrared. Because these multiple illumination requirements cannot be met with a single light source, manufacturers now offer adapters that enable two or more lamps to be simultaneously attached to a single microscope.
Fluorescence Filter Combinations
As previously discussed, light passing through the lenses and diaphragms of the vertical illuminator finally encounters the excitation filter housed in an optical block positioned to coincide with the axial intersection between the illuminator light path and the microscope optical train. The excitation filter selects a narrow band of wavelengths from the wide spectrum generated by the lamp and passes them to the dichromatic mirror, which in turn, reflects the light through the objective and onto the specimen. Fluorescence emission gathered by the objective passes once again through the dichromatic mirror and the emission or barrier filter before forming an image in the fixed eyepiece diaphragm. The common filters employed to isolate wavelength bands in widefield fluorescence microscopy include colored glass filters and interference thin film filters (or a combination of the two). Identifying the proper filters for each step in the fluorescence microscopy illumination and imaging scheme can be confusing due the large number of filter manufacturers involved, each of which contributes a proprietary alphanumeric nomenclature to the fluorescence literature.
The anatomy of a typical fluorescence filter block is diagrammed schematically in Figure 7, along with the associated spectral profiles of the dichromatic mirror, excitation, and barrier filters. Filter blocks are usually assembled with custom tools supplied by the manufacturer so the operator can interchange filters and the dichromatic mirror. The excitation and barrier filters are secured into place with retainer clips, optical glue, or circular threaded mounts (see Figure 7). In general, these filters can be removed without opening the optical block because they are positioned over recessed apertures on the flat exterior surfaces. Replacing a dichromatic mirror is more difficult and requires complete disassembly of the block to gain access to the interior. Most block sections are cast with a 45-degree diagonal joint that serves double duty by protecting the interior and supporting the dichromatic mirror at the proper angle. After removing the fasteners holding the block sections together (pins or small screws), the mirror can be removed by loosening or moving the retainer clip, and then very carefully dropping it out of the block. The dichromatic mirror should be handled with caution because the interference coating is usually not protected and can be easily scratched. Several of the filter manufacturers who supply the microscope companies also offer a wide selection of aftermarket filters and dichromatic mirrors for a variety of fluorescence applications.
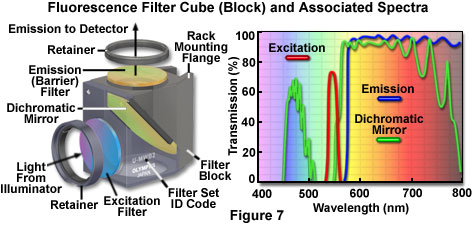
Fluorescence filter designs include longpass, shortpass (edge filters), and the narrow, medium, and wide family of bandpass filters. Examples of several common filter profiles are illustrated by the spectra presented in Figure 7. The emission filter spectrum (blue curve) in Figure 7 is produced by a longpass interference filter having a cut-on wavelength of approximately 575 nanometers. Longer wavelengths are transmitted through the filter, while shorter wavelengths are blocked. A narrow bandpass excitation filter from the same set (red curve, Figure 7) has a bandwidth of approximately 20 nanometers, while the dichromatic mirror (green curve) has transmission regions approximating a medium (455-490 nanometers) and wide bandpass filter (560-775 nanometers). Because the dichromatic mirror effectively serves as a longpass filter in the green, yellow and red regions of the visible spectrum (560 to 700 nanometers), it is treated as such in filter sets. A working knowledge of how fluorophore absorption and emission spectral profiles can be utilized to select the appropriate filter set for fluorescence microscopy is essential to successful implementation of this technique.
Dichromatic mirrors (or beamsplitters) are the most critical component in a fluorescence microscopy filter combination, and resemble longpass interference-type filters that are fabricated to close tolerances with multiple layers of dielectric materials. The major difference between a dichromatic mirror and a standard interference filter is that the mirror is specifically designed for reflection and transmission at defined boundary wavelengths, and must operate at a 45-degree angle with respect to the microscope and illuminator optical axes. Dichromatic mirrors are positioned with the interference coating facing the excitation light source in order to reflect short excitation wavelengths at a 90-degree angle through the optical train to the specimen. The same mirror must also act as a transmission filter to pass long wavelength fluorescence emission from the objective to the image plane. Because the wavelength transition region between almost total reflection and maximum transmission is often limited to only 20 or 30 nanometers, the dichromatic mirror is able to precisely discriminate between excitation and emission wavelengths.
Fluorescence filter sets are designed so that a particular band of excitation wavelengths exactly matches the reflection region in the dichromatic mirror. The result is efficient reflection of excitation light through the microscope and onto the specimen. Fluorescence emission from the specimen must match a high transmission region in the dichromatic mirror in order to enable these wavelengths to pass through to the detector. The barrier filter is less important in the overall scheme, but still plays an important role in ensuring that scattered and reflected excitation wavelengths, fluorescence from probes other than the target, and general background intensity due to autofluorescence is removed. The most important factor in creating a filter set is to make certain that the transmission, reflection, and emission profiles of the involved filters match in the appropriate regions. Otherwise, excitation wavelengths might be able to pass through the dichromatic mirror and fog the image, or fluorescence emission can inadvertently reflect at the mirror to compromise image brightness.
Even in seemingly perfectly matched filter combinations, slight overlaps between spectral profiles of the individual filters can occur to diminish performance. Of particular concern is crosstalk between the excitation filter and the dichromatic mirror, which enables some of the excitation light to pass through the mirror and reflect from the walls of the filter block. Light reflected at highly oblique angles can be partially transmitted through the barrier filter, as discussed above, to reduce image contrast. This type of leakage through filters is termed bleed-through or crossover, and occurs to a greater or lesser extent with practically all filter combinations. One of the primary areas of concern with both filter manufacturers and microscope companies is to improve the design of fluorescence filter combinations in order to reduce the level of crossover.

Several of the configurations for fluorescence filter sets are illustrated in Figure 8. The filter block turret (Figure 8(a)) contains five blocks, enabling simultaneous investigation of at least this number of fluorophores with rapid changeover between filter combinations. One of the turret slots is usually filled with a dummy optical block or left vacant for transmitted light observations. Fine-tuning of the excitation spectrum for imaging dual or multiply labeled specimens is accomplished with excitation balancers (Figure 8(b)), which contain a single interference filter mounted on a swivel. Rotating the adjustment lever of an excitation balancer shifts the bandpass transmission region of the filter to shorter wavelengths. Thus, when the balancer filter is rotated from an incident angle of zero degrees (perpendicular to the vertical illuminator axis) to the maximum swivel angle of 45 degrees, the bandpass region of the filter can be shifted by values ranging between 25 and 50 nanometers. Excitation balancers can be employed singly or in tandem to alter the excitation bandwidths of fluorochromes under observation in order to equalize the emission intensities. This feature enables fine-tuning of fluorophore intensity in specimens containing several probes to, for example, reduce the fluorescence emission from one probe while increasing the intensity of another.
The rapid advances in thin film coating technology are evidenced by the creation of multiple transmission peaks in a single interference filter and the ability to fabricate interleaved bands of reflection and transmission in dichromatic mirrors. When properly matched, two multi-band filters and a dichromatic mirror can be combined to produce a multiple fluorescence filter set that enables simultaneous excitation and observation of emission by several fluorophores. Filter sets are available from the manufacturers that are suitable for use with two, three, and even four fluorophores in the same specimen. The major problem with multiple filter sets is their expense and the excessive amount of crossover or bleed-through that occurs when emission from one of the fluorescent probes is transmitted through a bandpass region intended for another. In some specimens (and filter sets), bleed-through has a significant impact on background intensity and image contrast. Many investigators prefer to image each fluorophore separately with optimized filter sets, subsequently combining the images into a composite.
Advanced fluorescence techniques often require the use of several excitation and emission filters with a single dichromatic mirror. In order to facilitate these investigations, many fluorescence microscopes are equipped with motorized filter wheels or sliders containing up to six individual filters (see Figure 8(c)). Microscopes designed to be used with the sliders either contain a special slot in the vertical illuminator for insertion of the slider, or the filter slider can be substituted for one that normally contains neutral density filters. Emission sliders can also be accommodated by the vertical illuminator in some microscope designs, or an auxiliary intermediate tube installed between the illuminator and observation tubes to house the slider. Motorized filter wheel units containing excitation filters are generally sandwiched into the vertical illuminator optical pathway between the neutral density filters and the lamphouse in both upright and inverted microscopes. Likewise, the motorized emission filter wheel unit is placed between the vertical illuminator and the eyepiece observation tubes in upright microscope designs, but is usually attached through the same external port as the detector for inverted instruments. Excitation and emission sliders and motorized filter wheels are extremely useful for dual and triple excitation applications with a single dichromatic mirror. By eliminating the need to re-position filter blocks while observing a single specimen, these accessories enable the investigator to obtain accurate registration between images.
Objectives for Fluorescence Microscopy
In all forms of reflected light microscopy (including fluorescence), image intensity is a function of the objective numerical aperture and magnification. In fact, the intensity or brightness (defined as photon flux per unit area and time) increases by the fourth power of the numerical aperture, but is inversely proportional to only the square of the magnification:
where NA is the objective numerical aperture and M is the magnification. It is obvious from this relationship that the brightest fluorescence images will be gathered by an objective with high numerical aperture and low magnification (such as a 0.75/20x). As an example, a 60x plan apochromat oil immersion objective with a numerical aperture of 1.4 will theoretically produce brighter images than a 100x objective of the same numerical aperture, but there are trade-offs to consider. Increasing the number of internal lens elements (as with the highest numerical aperture objectives) produces corresponding increases in autofluorescence and intensity-reducing reflections from the internal lens surfaces. Often, a compromise between the highest correction factors and increased light transmission is made by the manufacturers in their recommended objectives for fluorescence microscopy.
In general, the high numerical aperture oil (1.3 to 1.4) and water (1.2) immersion plan fluorite and plan apochromatic objectives produce the brightest fluorescence images because of their tremendous light-gathering ability. These objectives exhibit excellent color correction, and thus, are able to focus a wide range of fluorescence emission wavelengths in the same plane. The light transmission characteristics of apochromatic and fluorite objectives are excellent down to about 350 nanometers, an absolute requirement for examining fluorochromes that are excited in the ultraviolet region (such as DAPI, Hoechst, Alexa Fluor-350, and AMC). Despite having numerous internal lenses, these objectives are fabricated from low-fluorescence glass with anti-reflection coatings to minimize background fluorescence and yield images of very high contrast. Image brightness during observation can also be increased by reducing the eyepiece magnification factor from the standard 10x down to 8x or lower.
Objectives designed for specialized applications are widely available for fluorescence microscopy. Included in this class are high numerical aperture water immersion, multi-immersion (oil, water, and glycerin), and ultraviolet objectives that have quartz lens elements. For live cell imaging applications, long working distance objectives with correction collars are necessary for viewing specimens through thick (0.5-2 millimeter) culture flask walls. Examinations deep within thick specimens are facilitated by objectives having a very long focal length (working distance), which are available from several manufacturers for imaging with cover glasses through air or water. Long working distance water immersion objectives designed to be used without a cover glass also have a Teflon nosecone so that the objective can be dipped into aqueous solutions. Similar objectives are produced for ultraviolet excitation (340 nanometers) in water at working distances between 20 and 30 millimeters (including approximately 5 millimeters of water). For relatively low magnification investigations, 20x water immersion objectives with very high numerical aperture (0.75 to 0.95) are recommended. Although quite expensive, these objectives produce extremely bright images in situations where fluorophore concentrations and/or quantum yields are marginal.
Accessories for Fluorescence Microscopy
Manufacturers are continually producing useful add-on accessory units for their instruments in order to increase the available options for the ever-growing number of imaging applications in fluorescence microscopy. For example, a rectangular field stop (see Figure 9), which restricts the specimen area being illuminated to improve contrast and reduce photobleaching, is available as an option on some microscopes. The modular field stop is substituted for the traditional iris field diaphragm (also a removable module) in the vertical illuminator and is designed to match the aspect ratio of digital imaging sensors. Rectangular field stops improve the efficiency of Köhler illumination and reduce the signal-to-noise ratio for electronic sensors. As an added benefit, the image rectangle size can be adjusted to coincide with stage step size when using programmable scanning stages in deconvolution investigations. Pinhole apertures, housed in modules similar to the rectangular field stop, are available for applications requiring a highly restricted illumination field.

Other accessories include double lamp housing adapters that enable two light sources (such as mercury and xenon arc-discharge lamps) to be simultaneously attached to the vertical illuminator. Specialized illuminators that adapt lasers for excitation are also available for applications such as total internal reflection fluorescence (TIRF), fluorescence lifetime imaging microscopy (FLIM), ratio imaging, and photobleaching recovery experiments. In addition, most of the research-level fluorescence microscopes offered by the major manufacturers are readily adapted to their respective laser scanning confocal accessories. Upright microscopes accept confocal scan units through the trinocular observation tube, while inverted instruments can attach the beam scanners to side or rear ports in the microscope frame. Dual ports that are inserted between the vertical illuminator and the observation tubes can be useful for both introducing additional light sources or directing fluorescence to more than one detector. Most of these units are available with optional C-mount adaptors or standard microscope port fittings. Dual port adapters are also available for simultaneously attaching a fiber optic cable from a laser source and an arc-discharge lamphouse to the rear of the vertical illuminator.
Fluorescence microscopes designed for electrophysiology investigations have become very sophisticated. Many are equipped with specialized vibration-dampened stages that can accept a wide variety of micromanipulator accessories for experiments using fluorescence, infrared differential interference contrast (IR-DIC), and traditional brightfield contrast-enhancement imaging techniques. Swinging nosepieces enable rapid vibration-free interchange of objectives to prevent disturbing specimens or the intrusion of air bubbles while imaging live cells and tissues. Fluorescence macro observation in living organisms is possible with new high numerical aperture long working distance 2x and 4x macro lenses (objectives), which are equipped with specialized filter combination blocks. In addition, several manufacturers offer fluorescence vertical illuminators and filter sets as accessories for their stereomicroscopes.
Magnification factors of 1.25x to 1.5x are sometimes available as an option on vertical illuminators, but the use of auxiliary lenses should be avoided whenever possible due to the inherent problems of introducing empty magnification into fluorescence images. Several modular vertical illuminator designs include provisions for attaching beamsplitter modules containing multiple camera ports above the illuminator to increase imaging capabilities. Considering the wide range of motorized accessories now available for fluorescence microscopes, including nosepieces, condensers, filter wheels, and stages, more sophisticated imaging techniques are available to investigators than ever before. The manufacturers exert a significant amount of effort in the design of accessories for many of the complex fluorescence applications that were once possible only with microscopes constructed using aftermarket parts from a variety of sources. As fluorescence microscopy becomes an increasingly critical tool in cell biology, neurophysiology, and the clinical arena, the development of innovative new microscope accessories will no doubt be a continuing trend.
Inverted Fluorescence Microscope Design
A similar version of the vertical fluorescence illuminator is available for inverted (tissue culture) microscope stands. The inverted stands also permit combining or alternating between reflected light fluorescence and the various contrast enhancing techniques of transmitted light microscopy. Research-level inverted microscopes feature multiple (up to six) input/output ports, usually with single ports on each side of the frame, as well as one or two ports (upper and lower) at the rear and a bottom port underneath the base of the microscope. In some models, primary images can be obtained simultaneously from three or more ports without the use of relay lenses. This level of connectivity enables the use of multiple light sources, filter wheels, and camera systems for complex fluorescence analysis. Mercury and xenon lamphouses for inverted microscopes are available with the standard multi-element or aspherical collector lenses to improve performance and reduce aberration in the ultraviolet and infrared spectral regions. In addition, a wide spectrum of lamphouse adapters can be utilized to attach several illumination sources, similar to the accessories available for upright microscopes.
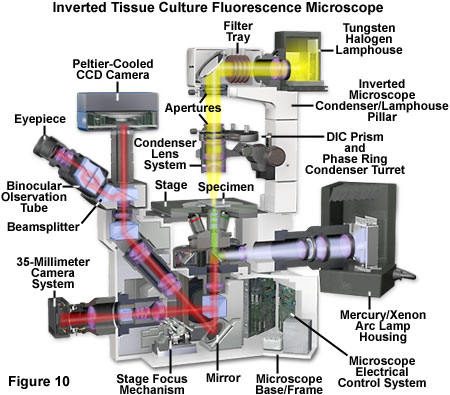
Presented in Figure 10 is a cut-away schematic diagram of a modern inverted (tissue culture) fluorescence microscope equipped with both a Peltier-cooled CCD image sensor and a traditional 35-millimeter film camera system. Although film cameras are of limited use, most inverted microscopes still include a port for these attachments in the lower portion of the front base. The microscope illustrated in Figure 10 can perform traditional brightfield transmitted illumination (with or without contrast enhancement) using the tungsten-halogen lamphouse mounted on the pillar. A mercury or xenon arc-discharge lamp is employed for fluorescence microscopy using a reflected light illuminator attached to a specially configured rear port. Aperture and field diaphragms, along with neutral density filters, are accessed near the port at the rear of the microscope. In some inverted microscope models (not illustrated), an L-shaped reflected light illuminator containing centerable diaphragms is available to improve access to the rear auxiliary ports for additional accessories. The light path through the microscope in Figure 10 is depicted in yellow for transmitted light, violet for unfiltered arc lamp illumination, green for filtered fluorescence excitation, and red for fluorescence emission.
Modern inverted microscope frames, like their upright counterparts, are computer engineered and fabricated with composite materials for structural and thermal stability. In addition, the mechanical stage components and circuit structures are designed with short travel distances and high rigidity to avoid pitch and yaw when the nosepiece is manipulated during routine operations such as DIC prism insertion or adjustment of objective correction collars. Advanced nosepiece stages are able to virtually eliminate focus drift during time-lapse and prolonged fluorescence observations. Other stage options not available for standard upright microscopes include gliding and 360-degree rotating stages, glass stage insert plates, heating plates, Petri dish and plate holders, and carbon dioxide incubator chambers.
Inverted microscopes having a modular design can easily be configured for investigations in electrophysiology, in vitro fertilization, micromanipulation, high-resolution DIC, video-enhanced observations, and a variety of advanced fluorescence techniques. The instruments are also readily adapted for confocal and multiphoton microscopy. Motorized accessories include shutters, filter wheels, revolving nosepieces, fluorescence block turrets, focus drives, and condensers. When coupled to the advanced objectives available in long working distance, water immersion, ultraviolet excitation, and phase contrast, all having a high degree of optical correction, inverted microscopes are ideal instruments for conducting fluorescence investigations on living cells and tissues.
Conclusions
In fluorescence microscopy, wide variations between localized fluorophore concentrations within the specimen, coupled to differences in extinction coefficient and quantum yield from one fluorochrome to another, significantly influence the emission signal produced for a given quantity of excitation intensity. Considering that many specimens contain only minute quantities of fluorescent material in any particular viewfield, these combined factors produce an average level of fluorescence emission that is four to six orders of magnitude less than the excitation intensity. In addition, several of the more sophisticated fluorescence techniques, such as in situ hybridization and resonance energy transfer (FRET), have emission signal intensities that can range nine to ten orders of magnitude less than that of the excitation. In order to compensate for these large discrepancies between the intensity of excitation and emission, modern fluorescence microscopes must be able to attenuate the excitation illumination by levels exceeding a billion times without disturbing the fluorescence signal.
Among the main attributes of fluorescence microscopy is the high specificity for fluorescent probes that absorb and emit light at characteristic wavelengths, leading to the ability of the technique to selectively detect a target species at very low concentrations in complex mixtures. In addition, the high sensitivity and spatial resolution of fluorescence enables single molecules to be pinpointed and studied at length scales beneath the optical resolution of the microscope. Localized environmental factors also weigh heavily on fluorescence emission, so the method is an ideal probe for fluctuations in pH, viscosity, ion concentrations, molecular distances and orientation, membrane potentials, hydrophobicity, charge distribution, and diffusion coefficients. The temporal resolution of fluorescence is limited to the lifetime of excited fluorescent probes, which can be on the order of nanoseconds. Because many biological processes occur in this time domain, decay kinetics can reveal dynamic information about cellular processes. Taken collectively, these factors are of critical importance in applications of fluorescence microscopy to cell biology.
Fluorescence microscopes have evolved with amazing speed over the past decade, coupled to equally rapid advances in laser technology, solid-state detectors, interference thin film fabrication, and computer-based image analysis. The development of high numerical aperture water immersion objectives has further assisted investigations of biological phenomena, allowing investigators to probe deep within the living cell in its natural environment. As the microscope manufacturers respond to the changing needs of the research community, the development of advanced fluorescence instruments and accessories will no doubt continue, and their ultimate contribution to explorations into nature's mysteries may ultimately be of profound significance.
Contributing Authors
Mortimer Abramowitz - Olympus America, Inc., Two Corporate Center Drive., Melville, New York, 11747.
Brian Herman - Department of Cellular and Structural Biology, University of Texas Health Science Center, 7703 Floyd Curl Drive, San Antonio, Texas 78229.
Douglas B. Murphy - Department of Cell Biology and Anatomy and Microscope Facility, Johns Hopkins University School of Medicine, 725 N. Wolfe Street, 107 WBSB, Baltimore, Maryland 21205.
Michael W. Davidson - National High Magnetic Field Laboratory, 1800 East Paul Dirac Dr., The Florida State University, Tallahassee, Florida, 32310.
BACK TO FLUORESCENCE MICROSCOPE ANATOMY
BACK TO FLUORESCENCE MICROSCOPY