
Introduction to Confocal Microscopy
Confocal microscopy offers several advantages over conventional widefield optical microscopy, including the ability to control depth of field, elimination or reduction of background information away from the focal plane (that leads to image degradation), and the capability to collect serial optical sections from thick specimens. The basic key to the confocal approach is the use of spatial filtering techniques to eliminate out-of-focus light or glare in specimens whose thickness exceeds the immediate plane of focus. There has been a tremendous explosion in the popularity of confocal microscopy in recent years, due in part to the relative ease with which extremely high-quality images can be obtained from specimens prepared for conventional fluorescence microscopy, and the growing number of applications in cell biology that rely on imaging both fixed and living cells and tissues. In fact, confocal technology is proving to be one of the most important advances ever achieved in optical microscopy.
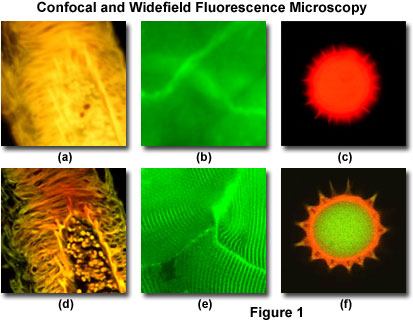
In a conventional widefield optical epi-fluorescence microscope, secondary fluorescence emitted by the specimen often occurs through the excited volume and obscures resolution of features that lie in the objective focal plane. The problem is compounded by thicker specimens (greater than 2 micrometers), which usually exhibit such a high degree of fluorescence emission that most of the fine detail is lost. Confocal microscopy provides only a marginal improvement in both axial (z; along the optical axis) and lateral (x and y; in the specimen plane) optical resolution, but is able to exclude secondary fluorescence in areas removed from the focal plane from resulting images. Even though resolution is somewhat enhanced with confocal microscopy over conventional widefield techniques, it is still considerably less than that of the transmission electron microscope. In this regard, confocal microscopy can be considered a bridge between these two classical methodologies.
Presented in Figure 1 are a series of images that compare selected viewfields in traditional widefield and laser scanning confocal fluorescence microscopy. A thick section of fluorescently stained human medulla in widefield fluorescence exhibits a large amount of glare from fluorescent structures above and below the focal plane (Figure 1(a)). When imaged with a laser scanning confocal microscope (Figure 1(d)), the medulla thick section reveals a significant degree of structural detail. Likewise, widefield fluorescence imaging of whole rabbit muscle fibers stained with fluorescein produce blurred images (Figure 1(b)) lacking in detail, while the same specimen field (Figure 1(e)) reveals a highly striated topography in confocal microscopy. Autofluorescence in a sunflower pollen grain produces an indistinct outline of the basic external morphology (Figure 1(c)), but yields no indication of the internal structure. In contrast, a thin optical section of the same grain (Figure 1(f)) acquired with confocal techniques displays a dramatic difference between the particle core and the surrounding envelope.
Historical Perspective
The basic concept of confocal microscopy was originally developed by Marvin Minsky in the mid-1950s (patented in 1957) when he was a postdoctoral student at Harvard University. Minsky wanted to image neural networks in unstained preparations of brain tissue and was driven by the desire to image biological events at they occur in living systems. Minsky's invention remained largely unnoticed, due most probably to the lack of intense light sources necessary for imaging and the computer horsepower required to handle large amounts of data. Following Minsky's work, M. David Egger and Mojmir Petran fabricated a multiple-beam confocal microscope in the late 1960s that utilized a spinning (Nipkow) disk for examining unstained brain sections and ganglion cells. Continuing in this arena, Egger went on to develop the first mechanically scanned confocal laser microscope, and published the first recognizable images of cells in 1973. During the late 1970s and the 1980s, advances in computer and laser technology, coupled to new algorithms for digital manipulation of images, led to a growing interest in confocal microscopy.
Fortuitously, shortly after Minsky's patent had expired, practical laser scanning confocal microscope designs were translated into working instruments by several investigators. Dutch physicist G. Fred Brakenhoff developed a scanning confocal microscope in 1979, while almost simultaneously, Colin Sheppard contributed to the technique with a theory of image formation. Tony Wilson, Brad Amos, and John White nurtured the concept and later (during the late 1980s) demonstrated the utility of confocal imaging in the examination of fluorescent biological specimens. The first commercial instruments appeared in 1987. During the 1990s, advances in optics and electronics afforded more stable and powerful lasers, high-efficiency scanning mirror units, high-throughput fiber optics, better thin film dielectric coatings, and detectors having reduced noise characteristics. In addition, fluorochromes that were more carefully matched to laser excitation lines were beginning to be synthesized. Coupled to the rapidly advancing computer processing speeds, enhanced displays, and large-volume storage technology emerging in the late 1990s, the stage was set for a virtual explosion in the number of applications that could be targeted with laser scanning confocal microscopy.
Modern confocal microscopes can be considered as completely integrated electronic systems where the optical microscope plays a central role in a configuration that consists of one or more electronic detectors, a computer (for image display, processing, output, and storage), and several laser systems combined with wavelength selection devices and a beam scanning assembly. In most cases, integration between the various components is so thorough that the entire confocal microscope is often collectively referred to as a digital or video imaging system capable of producing electronic images. These microscopes are now being employed for routine investigations on molecules, cells, and living tissues that were not possible just a few years ago.
Principles of Confocal Microscopy
The confocal principle in epi-fluorescence laser scanning microscope is diagrammatically presented in Figure 2. Coherent light emitted by the laser system (excitation source) passes through a pinhole aperture that is situated in a conjugate plane (confocal) with a scanning point on the specimen and a second pinhole aperture positioned in front of the detector (a photomultiplier tube). As the laser is reflected by a dichromatic mirror and scanned across the specimen in a defined focal plane, secondary fluorescence emitted from points on the specimen (in the same focal plane) pass back through the dichromatic mirror and are focused as a confocal point at the detector pinhole aperture.
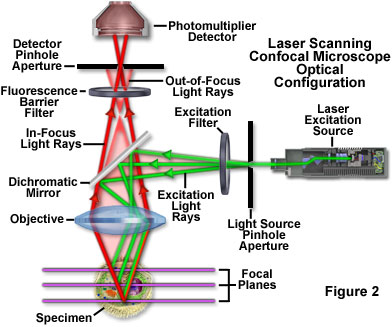
The significant amount of fluorescence emission that occurs at points above and below the objective focal plane is not confocal with the pinhole (termed Out-of-Focus Light Rays in Figure 2) and forms extended Airy disks in the aperture plane. Because only a small fraction of the out-of-focus fluorescence emission is delivered through the pinhole aperture, most of this extraneous light is not detected by the photomultiplier and does not contribute to the resulting image. The dichromatic mirror, barrier filter, and excitation filter perform similar functions to identical components in a widefield epi-fluorescence microscope. Refocusing the objective in a confocal microscope shifts the excitation and emission points on a specimen to a new plane that becomes confocal with the pinhole apertures of the light source and detector.
In traditional widefield epi-fluorescence microscopy, the entire specimen is subjected to intense illumination from an incoherent mercury or xenon arc-discharge lamp, and the resulting image of secondary fluorescence emission can be viewed directly in the eyepieces or projected onto the surface of an electronic array detector or traditional film plane. In contrast to this simple concept, the mechanism of image formation in a confocal microscope is fundamentally different. As discussed above, the confocal fluorescence microscope consists of multiple laser excitation sources, a scan head with optical and electronic components, electronic detectors (usually photomultipliers), and a computer for acquisition, processing, analysis, and display of images.
The scan head is at the heart of the confocal system and is responsible for rasterizing the excitation scans, as well as collecting the photon signals from the specimen that are required to assemble the final image. A typical scan had contains inputs from the external laser sources, fluorescence filter sets and dichromatic mirrors, a galvanometer-based raster scanning mirror system, variable pinhole apertures for generating the confocal image, and photomultiplier tube detectors tuned for different fluorescence wavelengths. The general arrangement of scan head components is presented in Figure 3 for a typical commercial unit.
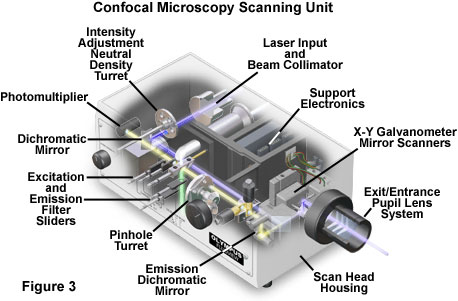
In epi-illumination scanning confocal microscopy, the laser light source and photomultiplier detectors are both separated from the specimen by the objective, which functions as a well-corrected condenser and objective combination. Internal fluorescence filter components (such as the excitation and barrier filters and the dichromatic mirrors) and neutral density filters are contained within the scanning unit (see Figure 3). Interference and neutral density filters are housed in rotating turrets or sliders that can be inserted into the light path by the operator. The excitation laser beam is connected to the scan unit with a fiber optic coupler followed by a beam expander that enables the thin laser beam wrist to completely fill the objective rear aperture (a critical requirement in confocal microscopy). Expanded laser light that passes through the microscope objective forms an intense diffraction-limited spot that is scanned by the coupled galvanometer mirrors in a raster pattern across the specimen plane (point scanning).
One of the most important components of the scanning unit is the pinhole aperture, which acts as a spatial filter at the conjugate image plane positioned directly in front of the photomultiplier. Several apertures of varying diameter are usually contained on a rotating turret that enables the operator to adjust pinhole size (and optical section thickness). Secondary fluorescence collected by the objective is descanned by the same galvanometer mirrors that form the raster pattern, and then passes through a barrier filter before reaching the pinhole aperture. The aperture serves to exclude fluorescence signals from out-of-focus features positioned above and below the focal plane, which are instead projected onto the aperture as Airy disks having a diameter much larger than those forming the image. These oversized disks are spread over a comparatively large area so that only a small fraction of light originating in planes away from the focal point passes through the aperture. The pinhole aperture also serves to eliminate much of the stray light passing through the optical system. Coupling of aperture-limited point scanning to a pinhole spatial filter at the conjugate image plane is an essential feature of the confocal microscope.
When contrasting the similarities and differences between widefield and confocal microscopes, it is often useful to compare the character and geometry of specimen illumination utilized for each of the techniques. Traditional widefield epi-fluorescence microscope objectives focus a wide cone of illumination over a large volume of the specimen, which is uniformly and simultaneously illuminated (as illustrated in Figure 4(a)). A majority of the fluorescence emission directed back towards the microscope is gathered by the objective (depending upon the numerical aperture) and projected into the eyepieces or detector. The result is a significant amount of signal due to emitted background light and autofluorescence originating from areas above and below the focal plane, which seriously reduces resolution and image contrast.

The laser illumination source in confocal microscopy is first expanded to fill the objective rear aperture, and then focused by the lens system to a very small spot at the focal plane (Figure 4(b)). The size of the illumination point ranges from approximately 0.25 to 0.8 micrometers in diameter (depending upon the objective numerical aperture) and 0.5 to 1.5 micrometers deep at the brightest intensity. Confocal spot size is determined by the microscope design, wavelength of incident laser light, objective characteristics, scanning unit settings, and the specimen. Presented in Figure 4 is a comparison between the typical illumination cones of a widefield (Figure 4(a)) and point scanning confocal (Figure 4(b)) microscope at the same numerical aperture. The entire depth of the specimen over a wide area is illuminated by the widefield microscope, while the sample is scanned with a finely focused spot of illumination that is centered in the focal plane in the confocal microscope.
In laser scanning confocal microscopy, the image of an extended specimen is generated by scanning the focused beam across a defined area in a raster pattern controlled by two high-speed oscillating mirrors driven by galvanometer motors. One of the mirrors moves the beam from left to right along the x lateral axis, while the other translates the beam in the y direction. After each single scan along the x axis, the beam is rapidly transported back to the starting point and shifted along the y axis to begin a new scan in a process termed flyback. During the flyback operation, image information is not collected. In this manner, the area of interest on the specimen in a single focal plane is excited by laser illumination from the scanning unit.
As each scan line passes along the specimen in the lateral focal plane, fluorescence emission is collected by the objective and passed back through the confocal optical system. The speed of the scanning mirrors is very slow relative to the speed of light, so the secondary emission follows a light path along the optical axis that is identical to the original excitation beam. Return of fluorescence emission through the galvanometer mirror system is referred to as descanning. After leaving the scanning mirrors, the fluorescence emission passes directly through the dichromatic mirror and is focused at the detector pinhole aperture. Unlike the raster scanning pattern of excitation light passing over the specimen, fluorescence emission remains in a steady position at the pinhole aperture, but fluctuates with respect to intensity over time as the illumination spot traverses the specimen producing variations in excitation.
Fluorescence emission that is passed through the pinhole aperture is converted into an analog electrical signal having a continuously varying voltage (corresponding to intensity) by the photomultiplier. The analog signal is periodically sampled and converted into pixels by an analog-to-digital (A/D) converter housed in the scanning unit or the accompanying electronics cabinet. The image information is temporarily stored in an image frame buffer card in the computer and displayed on the monitor. It is important to note that the confocal image of a specimen is reconstructed, point by point, from emission photon signals by the photomultiplier and accompanying electronics, yet never exists as a real image that can be observed through the microscope eyepieces.
Laser Scanning Confocal Microscope Configuration
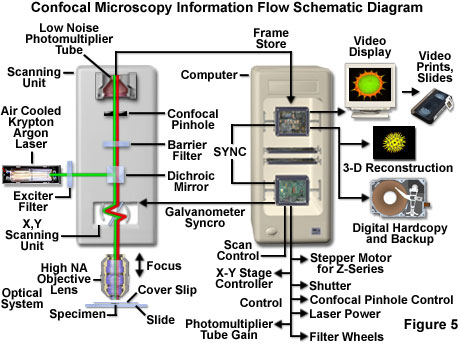
Basic microscope optical system characteristics have remained fundamentally unchanged for many decades due to engineering restrictions on objective design, the static properties of most specimens, and the fact that resolution is governed by the wavelength of light. However, fluorescent probes that are employed to add contrast to biological specimens and, and other technologies associated with optical microscopy techniques, have improved significantly. The explosive growth and development of the confocal approach is a direct result of a renaissance in optical microscopy that has been largely fueled by advances in modern optical and electronics technology. Among these are stable multi-wavelength laser systems that provide better coverage of the ultraviolet, visible, and near-infrared spectral regions, improved interference filters (including dichromatic mirrors, barrier, and excitation filters), sensitive low-noise wide band detectors, and far more powerful computers. The latter are now available with relatively low-cost memory arrays, image analysis software packages, high-resolution video displays, and high quality digital image printers. The flow of information through a modern confocal microscope is presented diagrammatically in Figure 5.
Although many of these technologies have been developed independently for a variety of specifically-targeted applications, they have been gradually been incorporated into mainstream commercial confocal microscopy systems. In current microscope systems, classification of designs is based on the technology utilized to scan specimens. Scanning can be accomplished either by translating the stage in the x, y, and z directions while the laser illumination spot is held in a fixed position, or the beam itself can be raster-scanned across the specimen. Because three-dimensional translation of the stage is cumbersome and prone to vibration, most modern instruments employ some type of beam-scanning mechanism.
In modern confocal microscopes, two fundamentally different techniques for beam scanning have been developed. Single-beam scanning, one of the more popular methods employed in a majority of the commercial laser scanning microscopes, uses a pair of computer-controlled galvanometer mirrors to scan the specimen in a raster pattern at a rate of approximately one frame per second. Faster scanning rates (to near video speed) can be achieved using acousto-optic devices or oscillating mirrors. In contrast, multiple-beam scanning confocal microscopes are equipped with a spinning Nipkow disk containing an array of pinholes and microlenses. These instruments often use arc-discharge lamps for illumination instead of lasers to reduce specimen damage and enhance the detection of low fluorescence levels during real time image collection. Another important feature of the multiple-beam microscopes is their ability to readily capture images with an array detector, such as a charge-coupled device (CCD) camera system.
All laser scanning confocal microscope designs are centered around a conventional upright or inverted research-level optical microscope. However, instead of the standard tungsten-halogen or mercury arc-discharge lamp, one or more laser systems are used as a light source to excite fluorophores in the specimen. Image information is gathered point by point with a specialized detector such as a photomultiplier tube or avalanche photodiode, and then digitized for processing by the host computer, which also controls the scanning mirrors and/or other devices to facilitate the collection and display of images. After a series of images (usually serial optical sections) has been acquired and stored on digital media, analysis can be conducted utilizing numerous image processing software packages available on the host or a secondary computer.
Advantages and Disadvantages of Confocal Microscopy
The primary advantage of laser scanning confocal microscopy is the ability to serially produce thin (0.5 to 1.5 micrometer) optical sections through fluorescent specimens that have a thickness ranging up to 50 micrometers or more. The image series is collected by coordinating incremental changes in the microscope fine focus mechanism (using a stepper motor) with sequential image acquisition at each step. Image information is restricted to a well-defined plane, rather than being complicated by signals arising from remote locations in the specimen. Contrast and definition are dramatically improved over widefield techniques due to the reduction in background fluorescence and improved signal-to-noise. Furthermore, optical sectioning eliminates artifacts that occur during physical sectioning and fluorescent staining of tissue specimens for traditional forms of microscopy. The non-invasive confocal optical sectioning technique enables the examination of both living and fixed specimens under a variety of conditions with enhanced clarity.
With most confocal microscopy software packages, optical sections are not restricted to the perpendicular lateral (x-y) plane, but can also be collected and displayed in transverse planes. Vertical sections in the x-z and y-z planes (parallel to the microscope optical axis) can be readily generated by most confocal software programs. Thus, the specimen appears as if it had been sectioned in a plane that is perpendicular to the lateral axis. In practice, vertical sections are obtained by combining a series of x-y scans taken along the z axis with the software, and then projecting a view of fluorescence intensity as it would appear should the microscope hardware have been capable of physically performing a vertical section.
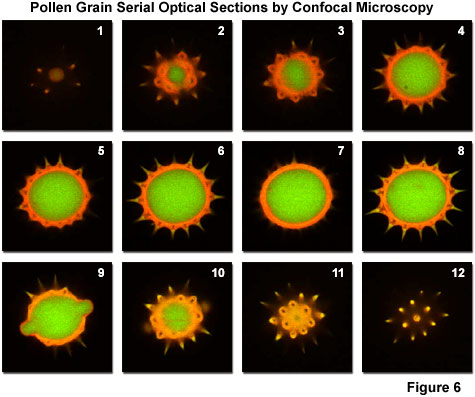
A typical stack of optical sections (often termed a z-series) through a sunflower pollen grain revealing internal variations in autofluorescence emission wavelengths are illustrated in Figure 6. Optical sections were gathered in 0.5-micrometer steps perpendicular to the z-axis (microscope optical axis) using a dual argon-ion (488 nanometer; green fluorescence) and green helium/neon (543 nanometer; red fluorescence) laser system. Pollen grains of from this species range between 20 and 40 micrometers in diameter and yield blurred images in widefield fluorescence microscopy (see Figure 1 (c)), which lack information about internal structural details. Although only 12 of the over 48 images collected through this series are presented in the figure, they represent individual focal planes separated by a distance of approximately 3 micrometers and provide a good indication of the internal grain structure.
In specimens more complex than a pollen grain, complex interconnected structural elements can be difficult to discern from a large series of optical sections sequentially acquired through the volume of a specimen with a laser scanning confocal microscope. However, once an adequate series of optical sections has been gathered, it can be further processed into a three-dimensional representation of the specimen using volume-rendering computational techniques. This approach is now in common use to help elucidate the numerous interrelationships between structure and function of cells and tissues in biological investigations. In order to ensure that adequate data is collected to produce a representative volume image, the optical sections should be recorded at the appropriate axial (z-step) intervals so that the actual depth of the specimen is reflected in the image.
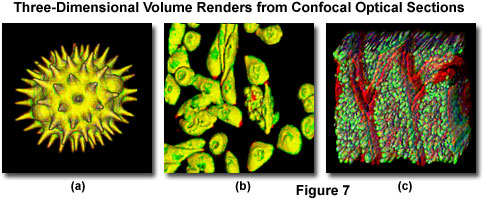
Most of the software packages accompanying commercial confocal instruments are capable of generating composite and multi-dimensional views of optical section data acquired from z-series image stacks. The three-dimensional software packages can be employed to create either a single three-dimensional representation of the specimen (Figure 7) or a video (movie) sequence compiled form different views of the specimen volume. These sequences often mimic the effect of rotation or similar spatial transformation that enhances the appreciation of the specimen's three-dimensional character. In addition, many software packages enable investigators to conduct measurements of length, volume, and depth, and specific parameters of the images, such as opacity, can be interactively altered to reveal internal structures of interest at differing levels within the specimen.
Typical three-dimensional representations of several specimens examined by serial optical sectioning are presented in Figure 7. The pollen grain optical sections illustrated in Figures 1 and 6 were combined to produce a realistic view of the exterior surface (Figure 7(a)) as it might appear if being examined by a scanning electron microscope. The algorithm utilized to construct the three-dimensional model enables the user to rotate the pollen grain through 360 degrees for examination. The tissue culture cells in Figure 7(b) are derived from the Chinese hamster ovary (CHO) line and were transfected with a chimeric plasmid vector containing the green fluorescent protein and a human immunodeficiency virus (HIV) protein that is expressed in the nucleus (thus, labeling the nuclear region). Thick tissue sections are also easily viewed in three-dimensions constructed from optical sections. The mouse intestine section illustrated in Figure 7(c) was labeled with several fluorophores and created from a stack of 45 optical sections.
In many cases, a composite or projection view produced from a series of optical sections provides important information about a three-dimensional specimen than a multi-dimensional view. For example, a fluorescently labeled neuron having numerous thin, extended processes in a tissue section is difficult (if not impossible) to image using widefield techniques due to out-of-focus blur. Confocal thin sections of the same neuron each reveal portions of several extensions, but these usually appear as fragmented streaks and dots and lack continuity. Composite views created by flattening a series of optical sections from the neuron will reveal all of the extended processes in sharp focus with well-defined continuity. Structural and functional analysis of other cell and tissue sections also benefits from composite views as opposed to, or coupled with, three-dimensional volume rendering techniques.
Advances in confocal microscopy have made possible multi-dimensional views of living cells and tissues that include image information in the x, y, and z dimensions as a function of time and presented in multiple colors (using two or more fluorophores). After volume processing of individual image stacks, the resulting data can be displayed as three-dimensional multicolor video sequences in real time. Note that unlike conventional widefield microscopy, all fluorochromes in multiply labeled specimens appear in register using the confocal microscope. Temporal data can be collected either from time-lapse experiments conducted over extended periods or through real time image acquisition in smaller frames for short periods of time. The potential for using multi-dimensional confocal microscopy as a powerful tool in cellular biology is continuing to grow as new laser systems are developed to limit cell damage and computer processing speeds and storage capacity improves.
Additional advantages of scanning confocal microscopy include the ability to adjust magnification electronically by varying the area scanned by the laser without having to change objectives. This feature is termed the zoom factor, and is usually employed to adjust the image spatial resolution by altering the scanning laser sampling period. Increasing the zoom factor reduces the specimen area scanned and simultaneously reduces the scanning rate. The result is an increased number of samples along a comparable length, which increases both the image spatial resolution and display magnification on the host computer monitor. Confocal zoom is typically employed to match digital image resolution with the optical resolution of the microscope when low numerical aperture and magnification objectives are being used to collect data.
Digitization of the sequential analog image data collected by the confocal microscope photomultiplier (or similar detector) facilitates computer image processing algorithms by transforming the continuous voltage stream into discrete digital increments that correspond to variations in light intensity. In addition to the benefits and speed that accrue from processing digital data, images can be readily prepared for print output or publication. In carefully controlled experiments, quantitative measurements of spatial fluorescence intensity (either statically or as a function of time) can also be obtained from the digital data.
Disadvantages of confocal microscopy are limited primarily to the limited number of excitation wavelengths available with common lasers (referred to as laser lines), which occur over very narrow bands and are expensive to produce in the ultraviolet region. In contrast, conventional widefield microscopes use mercury or xenon based arc-discharge lamps to provide a full range of excitation wavelengths in the ultraviolet, visible, and near-infrared spectral regions. Another downside is the harmful nature of high-intensity laser irradiation to living cells and tissues (an issue that has recently been addressed by multiphoton and Nipkow disk confocal imaging). Finally, the high cost of purchasing and operating multi-user confocal microscope systems, which can range up to an order of magnitude higher than comparable widefield microscopes, often limits their implementation in smaller laboratories. This problem can be easily overcome by cost-shared microscope systems that service one or more departments in a core facility. The recent introduction of personal confocal systems has competitively driven down the price of low-end confocal microscopes and increased the number of individual users.
Contributing Authors
Stephen W. Paddock - Laboratory of Molecular Biology, Howard Hughes Medical Institute, University of Wisconsin, Madison, Wisconsin 53706.
James B. Pawley - Department of Zoology, 1117 W. Johnson Dr., University of Wisconsin, Madison, Wisconsin 53706.
Thomas J. Fellers, and Michael W. Davidson - National High Magnetic Field Laboratory, 1800 East Paul Dirac Dr., The Florida State University, Tallahassee, Florida, 32310.