
Fluorescence
Overview of Excitation and Emission Fundamentals
Because of their novel electronic configurations, fluorochromes have unique and characteristic spectra for absorption (usually similar to excitation) and emission. These absorption and emission spectra show relative Intensity of fluorescence, with the relative intensity classically plotted on the vertical axis versus wavelength on the horizontal axis. For a given fluorochrome, the manufacturers indicate the wavelength for the peak of the illumination excitation intensity and the wavelength for the peak of fluorescence emission intensity. It is important to understand the origin of the graphs and curves displaying the excitation and emission spectra for a given fluorochrome.
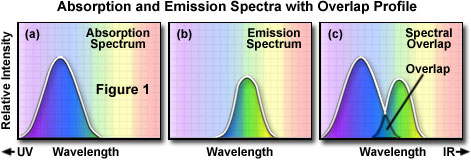
In order to determine the emission spectrum of a particular fluorochrome, the wavelength of maximum absorption (usually the same as the excitation maximum) is determined and the fluorochrome is excited at that wavelength. The absorption spectrum of a typical fluorochrome is illustrated in Figure 1(a) where the relative intensity of absorption is plotted against the measured wavelength. A monochromator (a device that allows narrow bands of light wavelengths to pass) is then used to scan the fluorescence emission intensity over the entire series of emission wavelengths. The relative intensity of the fluorescence is measured at the various wavelengths to plot the emission spectrum, as illustrated in Figure 1(b). The excitation spectrum of a given fluorochrome is determined in a similar manner by monitoring fluorescence emission at the wavelength of maximum intensity while the fluorophore is excited through a group consecutive wavelengths. The emission maximum is chosen and only emission light at that wavelength is allowed to pass to the detector. Excitation is induced (usually by means of a monochromator) at various excitation wavelengths and the intensity of the emitted fluorescence is measured as a function of wavelength. The result is a graph or curve (illustrated in Figure 1(a)), which depicts the relative fluorescence intensity produced by excitation over the spectrum of excitation wavelengths.
| Interactive Tutorial | |||||||||||
|
|||||||||||
Several observations can be made from a typical excitation and emission set of curves or spectra. There is usually an overlap between the higher wavelength end of the excitation spectrum and the lower wavelength end of the emission spectrum. This overlap of excitation and emission intensities and wavelengths (illustrated in Figure 1(c)) must be eliminated, in fluorescence microscopy, by means of the appropriate selection for an excitation filter, dichromatic beamsplitter (in reflected light fluorescence), and barrier or emission filter. Otherwise, the much brighter excitation light overwhelms the weaker emitted fluorescence light and significantly diminishes specimen contrast.
When electrons go from the excited state to the ground state (see the section below entitled Molecular Explanation), there is a loss of vibrational energy. As a result, the emission spectrum is shifted to longer wavelengths than the excitation spectrum (wavelength varies inversely to radiation energy). This phenomenon is known as Stokes Law or Stokes shift. The greater the Stokes shift, the easier it is to separate excitation light from emission light. The emission intensity peak is usually lower than the excitation peak, and the emission curve is often a mirror image of the excitation curve, but shifted to longer wavelengths. In order to achieve maximum fluorescence intensity, the fluorochrome is usually excited at the wavelength at the peak of the excitation curve, and the emission detection is selected at the peak wavelength (or other wavelengths chosen by the observer) of the emission curve. The selections of excitation wavelengths and emission wavelengths are controlled by appropriate filters. In determining the spectral response of an optical system, technical corrections are required to take into account such factors as glass transmission and detector sensitivity variables for different wavelengths.
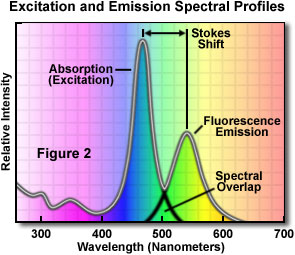
A typical fluorochrome absorption-emission spectral diagram is illustrated in Figure 2. Note that the curves of fluorescence intensity for absorption (usually similar to the excitation curve for pure compounds) and emission for this typical fluorochrome are somewhat similar in shape. The wavelength shift between excitation and emission has been known since the middle of the nineteenth century (Stokes Law). Also note that the excitation and emission curves overlap somewhat at the upper end of the excitation and the lower wavelengths of the emission curve.
| Interactive Tutorial | |||||||||||
|
|||||||||||
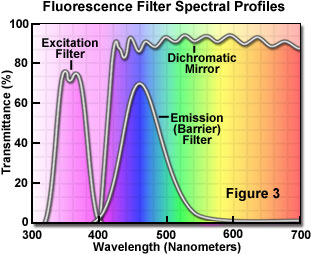
The separation of excitation and emission wavelengths is achieved by the proper selection of filters to block or pass specific wavelengths of the spectrum as presented in Figure 3. The design of fluorescence illuminators is based on control of excitation light and emission light by readily changeable filter insertions into the light path on the way toward the specimen and then emanating from the specimen. It is important, in view of low emission intensities, that the light source chosen for excitation be of sufficient brightness so that the relatively weak emission light can be maximized, and that fluorochromes of satisfactory absorption and yield be chosen.
The efficiency with which the fluorochrome absorbs the excitation light is known as the extinction coefficient. The greater the extinction coefficient, the greater the possibility of light absorption in a given wavelength region (a prerequisite to ensuing fluorescence emission). The yield of emitted light is referred to as the quantum yield, the ratio of the number of quanta ("packets" of energy) emitted compared to the number of quanta absorbed (usually the yield is between 0.1 and 0.9). Quantum yields below 1 are the result of the loss of energy through non-radiative pathways (such as heat or a photochemical reaction) rather than the re-radiative pathway of fluorescence. Listed below in Table 1 are fluorescence quantum yields for a group of selected fluorochromes. Notice that some quantum yields seem trivial (benzene) while others are very efficient (Fluorescein and Rhodamine-B).
Fluorochrome Fluorescence Quantum Yields
|
|||||||||||||||||||||||||||||||||||||
Table 1
Extinction coefficient, quantum yield, mean luminous intensity of the light source, and fluorescence lifetime are all important factors contributing to the intensity and utility of fluorescence emission. In addition, the localized environment surrounding the fluorochrome plays a paramount role in determining the characteristics of fluorescence emission. Variables such as solvent viscosity, ionic concentrations, pH, and hydrophobicity in the environment can have profound effects on both the fluorescence intensity and the lifetime of the excited state.
Molecular Explanation of Fluorescence
Fluorescence activity is sometimes depicted diagrammatically as shown in Figure 4(a) (termed a Jablonski energy diagram). Prior to excitation, the electronic configuration of the molecule is described as being in the ground state. Upon absorbing a photon of excitation light, usually of short wavelengths, electrons may be raised to a higher energy and vibrational excited state, a process that may only take a quadrillionth of a second (a time period commonly referred to as a femtosecond, 10E-15 seconds).
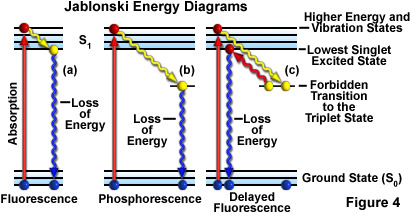
In fluorescence, during an interval of approximately a trillionth of a second (a picosecond or 10E-12 seconds), the excited electrons may lose some vibrational energy to the surrounding environment and return to what is called the lowest excited singlet state. From the lowest excited singlet state, the electrons are then able to "relax" back to the ground state with simultaneous emission of fluorescent light as illustrated in Figure 4(a). The emitted light is always of longer wavelength than the excitation light (Stokes Law) and continues so long as the excitation illumination bathes the fluorescent specimen. If the exciting radiation is halted, the fluorescence ceases.
| Interactive Tutorial | |||||||||||
|
|||||||||||
Occasionally the excited electrons, instead of relaxing to the lowest singlet state through vibrational interactions, make a forbidden transition to the exited triplet state and then to the ground state in a process where the emission of radiation may be considerably delayed�up to several seconds or more. This phenomenon is characteristic of phosphorescence, as shown in Figure 4(b). In some instances, the excited electrons may go from the triplet state back to the lowest excited singlet state and then return to the ground state, subsequently emitting fluorescent light. This action takes a little longer (about a microsecond or two) than usual fluorescence and is called delayed fluorescence (Figure 4(c)). Under other circumstances (for example, photobleaching or the presence of salts of heavy metals or other chemicals), emitted light may be significantly reduced or halted altogether, as discussed below.
Fading or Photobleaching
There are specific conditions that may affect the re-radiation of light by an excited fluorophore, and thus reduce the intensity of fluorescence. This reduction of emission intensity is generally called fading or photobleaching. Some authors further subdivide fading into quenching and bleaching. Bleaching is irreversible decomposition of the fluorescent molecules because of light intensity in the presence of molecular oxygen. Quenching also results in reduced fluorescence intensity and frequently is brought about as a result of oxidizing agents or the presence of salts of heavy metals or halogen compounds.
Often, quenching results from the transfer of energy to other acceptor molecules physically close to the excited fluorophores, a phenomenon known as resonance energy transfer. This particular phenomenon has become the basis for a newer technique of measuring distances far below the lateral resolution of the light microscope.
The occurrence of bleaching has led to a technique known as FRAP, or fluorescence recovery after photobleaching. FRAP is based upon bleaching by short laser bursts and subsequent observation of the recovery of fluorescence caused by the diffusion of fluorophores into the bleached area.
Properties of Popular Antifade Reagents
|
||||||||||||
Table 2
To reduce the degree of fading in some specimens, it may be advisable to use a neutral density filter in the light path before the illumination reaches the excitation filter, thus diminishing the excitation light intensity. In other instances, fading effects may be reduced by changing the pH of the mounting medium or by using anti-bleaching agents (several of the more important agents are listed in Table 2). For digital imaging, photomicrography, or simply visual observation, rapidly changing the field of view may also avoid fading effects.
Contributing Authors
Mortimer Abramowitz - Olympus America, Inc., Two Corporate Center Drive., Melville, New York, 11747.
Michael W. Davidson - National High Magnetic Field Laboratory, 1800 East Paul Dirac Dr., The Florida State University, Tallahassee, Florida, 32310.
BACK TO FLUORESCENCE INTRODUCTION